Болезнь Фабри в практике ревматолога
Болезнь Фабри – это редкое наследственное заболевание, характеризующееся нарушением разрушения и накоплением гликофосфолипидов в лизосомах клеток различных органов в результате дефицита лизосомного фермента α-галактозидазы А. Первые симптомы болезни Фабри (нейропатическая боль, ангиокератомы, желудочно-кишечные нарушения, снижение или отсутствие потоотделения) обычно появляются в детском или подростковом возрасте. Неблагоприятные исходы заболевания связаны с поражением почек, центральной нервной системы и сердца, развивающимся в возрасте 20-40 лет. Важное значение для диагностики болезни Фабри, позволяющей своевременно начать ферментозаместительную терапию, имеет информированность врачей, в том числе ревматологов, о проявлениях этого заболевания.
Болезнь Фабри – это прогрессирующее наследственное заболевание, сцепленное с Х-хромосомой, которое характеризуется накоплением гликофосфолипидов в лизосомах клеток различных органов в результате недостаточности или отсутствия a-галактозидазы А. Причиной дефицита этого фермента являются мутации гена GLA, расположенного на Х-хромосоме. Распространенность болезни Фабри в общей популяции составляет 50-100 на 1 млн населения и сопоставима с таковой АНЦА-ассоциированных васкулитов [1], а результаты скрининговых исследований среди новорожденных свидетельствуют о том, что распространенность этого заболевания может быть еще выше [2].
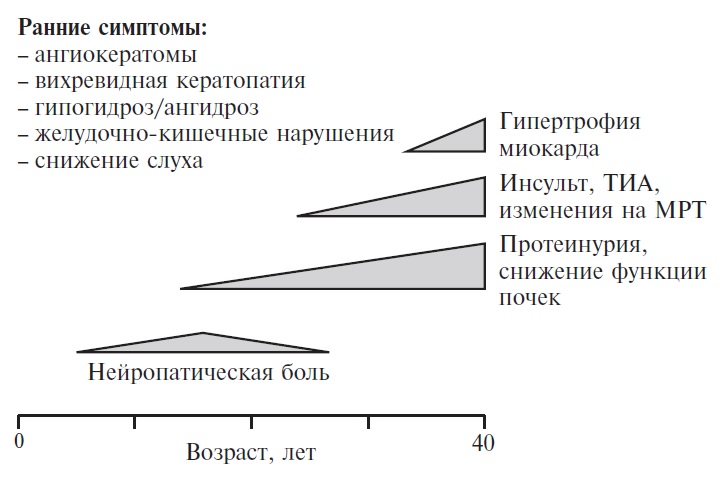
У большинства пациентов с болезнью Фабри первые симптомы, в частности нейропатическая боль, ангиокератомы, снижение или отсутствие потоотделения, желудочно-кишечные нарушения, появляются в детском или подростковом возрасте, а в возрасте 20-40 лет развивается поражение внутренних органов, в том числе почек (протеинурия и прогрессирующее снижение скорости клубочковой фильтрации), сердца (гипертрофия миокарда) и центральной нервной системы (транзиторные ишемические атаки и инсульт) (рис. 1). Заподозрить диагноз обычно позволяют начало болезни в детском возрасте, "классические" симптомы, системность поражения внутренних органов, наличие сходных или других проявлений болезни Фабри у родственников пациента. Однако даже при наличии типичных симптомов диагноз болезни Фабри часто устанавливают с большим опозданием, что отражает низкую информированность врачей об этом заболевании. По данным регистра Fabry Outcome Survey (FOS), в Европе и других странах мира за последние годы срок между появлением симптомов и установлением диагноза у взрослых пациентов несколько сократился, однако по-прежнему остается большим (медиана – 10,5 лет) [3]. В 2013-2017 гг. в клинике им. Е.М. Тареева было обследовано 82 взрослых пациента с болезнью Фабри. Медиана срока от первых симптомов до установления диагноза составила 18 (9; 27) лет, а диапазон варьировался от 0 до 51 года, причем у большинства пациентов диагноз не был заподозрен врачами, а установлен при скрининге, проводившемся в диализных отделениях, или в результате обследования родственников больных, т.е. семейного скрининга. Важность своевременной диагностики определяется возможностью заместительной терапии рекомбинантными препаратами a-галактозидазы А, которая позволяет уменьшить клинические симптомы, улучшить качество жизни и задержать прогрессирование поражения внутренних органов [4,5].
Учитывая многообразие клинических проявлений болезни Фабри, пациенты могут обращаться к врачам различных специальностей, в том числе терапевтам, неврологам, кардиологам, нефрологам, дерматологам, генетикам, а также ревматологам. Например, в нашей когорте по крайней мере один "ревматологический" диагноз был установлен у 22 (26,8%) из 82 пациентов (табл. 1).
| Диагноз | n (%) |
| Примечание: *васкулиты включали в себя IgA-васкулит, болезнь Бехчета и др., **артрит включал в себя ревматоидный артрит, ювенильный ревматоидный артрит и остеоартрит | |
| Васкулит* | 6 (7,3%) |
| Артрит** | 5 (6,1%) |
| Болезнь Ослера-Вебера-Рандю | 4 (4,9%) |
| Ревматическая лихорадка | 4 (4,9%) |
| Системная красная волчанка | 3 (3,7%) |
| Периодическая болезнь | 1 (1,2%) |
Болезнь Фабри как аутовоспалительное заболевание
В основе развития болезни Фабри лежат нарушение разрушения гликосфинголипидов и их отложение в различных органах и тканях, однако накопление субстратов a-галактозидазы А не позволяет объяснить некоторые клинические проявления и особенности течения болезни у части пациентов, в частности возможность ее прогрессирования несмотря на ферментозаместительную терапию. Высказано предположение, что определенный вклад в патогенез болезни Фабри вносит аутовоспаление. Этот термин был предложен в конце XX века M.McDermott и соавт., которые описали группу пациентов с TRAPS-синдромом (семейной периодической лихорадкой, ассоциированной с патологией рецептора к фактору некроза опухоли-a) [6]. В отличие от аутоиммунного воспаления, в развитии аутовоспалительных болезней главную роль играют генетически детерминированные реакции иммунитета и воспаление, а не антиген-индуцированные синтез антител или активация Т-лимфоцитов [7]. Соответственно, при обследовании у пациентов выявляют высокие лабораторные показатели активности воспаления при отсутствии аутоантител или признаков активации иммунных клеток, а причиной повреждения тканей являются про воспалительные цитокины, хемокины и молекулы адгезии.
Пусковым механизмом аутовоспаления является образование инфламмасомы (от англ. inflammation – воспаление) – макромолекулярного комплекса, в состав которого входят NLRP3 (криопирин) или другие родственные белки, а также белок ASC и прокаспаза-1, превращающаяся в активную каспазу-1. Последняя вызывает секрецию интерлейкина (ИЛ)-1β и ИЛ-18, которые, в свою очередь, индуцируют выделение ИЛ-6, ИЛ-8 и фактора некроза опухоли-a. Индукторами воспалительного ответа при аутовоспалительных заболеваниях могут служить молекулярные фрагменты, ассоциированные с повреждением (damage-associated molecular patterns – DAMP), – эндогенные молекулы, которые высвобождаются поврежденными клетками и распознаются специфическими рецепторами иммунных клеток.
В экспериментальных исследованиях добавление глоботриаозилцерамида к неизмененным клеткам вызывало апоптоз и секрецию цитокинов [8]. В связи с этим высказано предположение, что гликосфинголипиды, откладывающиеся в лизосомах, могут выступать в роли DAMP или индуцировать их образование в поврежденных клетках с последующим развитием хронического воспалительного ответа [9]. Подтверждением этой гипотезы может служить повышение сывороточных уровней провоспалительных цитокинов, в том числе ИЛ-1β, ИЛ-6, ФНО-α, моноцитарного хемоаттрактантного белка-1 (MCP-1), молекул адгезии у пациентов с болезнью Фабри и их снижение под действием ферментозаместительной терапии [10].
Концепция аутовоспаления может служить дополнительным доводом в пользу ранней ферментозаместительной терапии, так как персистирующее хроническое воспаление, вызванное отложением гликосфинголипидов, может способствовать развитию необратимых изменений в органах и тканях. Кроме того, аутовоспаление может быть причиной некоторых проявлений болезни Фабри, таких как преходящие лихорадка или артрит, характерных для ревматологических заболеваний.
Ревматологические маски болезни Фабри
Типичным симптомом болезни Фабри являются ангиокератомы – мелкие темно-красные мягкие узелки, локализующиеся на передней брюшной стенке, в частности внутри или вокруг пупка, в паховой области, на ягодицах, верхних конечностях, губах (рис. 2). Ангиокератомы наблюдались у 37 (45,7%) из 82 обследованных нами пациентов с болезнью Фабри. У большинства больных определялись распространенные кожные высыпания, однако в некоторых случаях ангиокератомы были единичными и располагались внутри пупка, на слизистой оболочке полости рта или половых губах. Практически у всех пациентов ангиокератомы появлялись в детском или подростковом возрасте и не уменьшались со временем. Многочисленные ангиокератомы могут быть расценены как пурпура ШенлейнаГеноха, что демонстрирует следующее наблюдение.
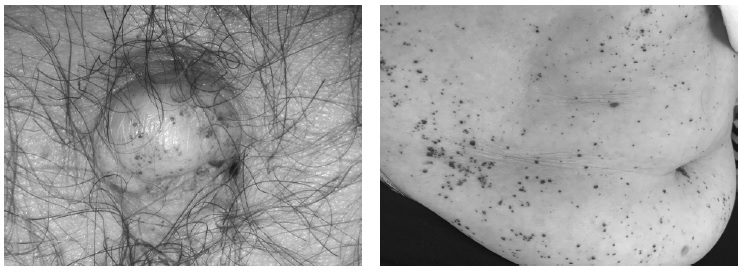
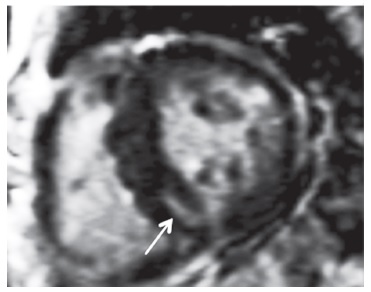
В 2013 году к нам в клинику был госпитализирован 30-летний мужчина, который с детства жаловался на боли в животе и ярко-красные высыпания на коже живота, ягодиц и нижних конечностей. Наблюдался ревматологом по месту жительства по поводу геморрагического васкулита. В возрасте 28 лет выявлена протеинурия без изменений мочевого осадка, в связи с чем получал лечение глюкокортикостероидами и циклофосфамидом без эффекта. При обследовании в клинике диагноз IgA-васкулита вызвал сомнения. Для этого заболевания характерны преходящие кожные высыпания, которые обычно появляются под действием физической нагрузки или холода и могут бесследно проходить в течение нескольких дней. Лечение глюкокортикостероидами обычно вызывает уменьшение кожного синдрома. В то же время у пациента кожные высыпания были постоянными и не уменьшились под влиянием глюкокортикостероидов. Кроме того, при обследовании, помимо поражения почек, была выявлена гипертрофия миокарда. При IgA-васкулите сердце не поражается, а увеличение массы миокарда левого желудочка отмечается только при наличии артериальной гипертонии, которая у пациента отсутствовала. Гипертрофия левого желудочка – это типичное проявление поражения сердца при болезни Фабри. При магнитно-резонансной томографии мы выявили гипертрофию левого желудочка у 16 (40,0%) из 40 взрослых пациентов с этим заболеванием, в том числе у 13 (46,4%) из 28 мужчин и 3 (25,0%) из 12 женщин (рис. 3) [11]. Следует отметить, что у трети пациентов наблюдалось асимметричное утолщение межжелудочковой перегородки, которое считают характерным для гипертрофической кардиомиопатии. Поражение почек развивается как при IgA-васкулите, так и при болезни Фабри, однако у пациентов с пурпурой ШенлейнаГеноха оно обычно проявляется не только небольшой протеинурией, но и микро- или макрогематурией, которая при болезни Фабри отсутствует. Учитывая сочетание ангиокератом и болей в животе, появившихся в детском возрасте, небольшой протеинурии, не сопровождавшейся изменением мочевого осадка, и гипертрофии миокарда, не связанной с артериальной гипертонией, был заподозрен диагноз болезни Фабри, который был подтвержден при молекулярно-генетическом исследовании. В течение 3 лет получает ферментозаместительную терапию. Протеинурия не нарастает. Функция почек остается нормальной.
К числу ранних симптомов болезни Фабри относится нейропатическая боль (акропарестезии), которая наблюдалась у 65 (79,3%) из 82 больных. У мальчиков она обычно появляется раньше, чем у девочек. У части пациентов нейропатическая боль имеет кризовое течение и характеризуется эпизодами мучительной жгучей боли в конечностях, которые возникают в жаркую погоду или при быстрой смене температуры окружающей среды или провоцируются лихорадкой, физической нагрузкой или стрессом. Некоторые пациенты пытаются уменьшить боль, опуская кисти и/или стопы в холодную воду. При болезни Фабри могут наблюдаться и практически постоянные боли и парестезии в кистях и стопах, хотя они, как правило, также усиливаются под действием выше перечисленных факторов. С возрастом нейропатическая боль иногда уменьшается или полностью проходит, что необходимо учитывать при сборе анамнеза у взрослого пациента. Иногда нейропатическую боль неправильно трактуют как боль в суставах, что может быть причиной ошибочных диагнозов, в том числе ревматоидного артрита.
Летом 2016 года в клинику был госпитализирован 20-летний мужчина, которого с 11 лет беспокоили приступы интенсивных болей в пальцах кистей и стоп, возникавшие 1-2 раза в неделю, продолжавшиеся 2-3 часа и проходившие после приема анальгетиков. Боли не сопровождались припуханием суставов. Тем не менее, на протяжении нескольких лет наблюдался педиатром по месту жительства с диагнозом ювенильного ревматоидного артрита. Принимал различные нестероидные противовоспалительные препараты (нимесулид, диклофенак, ацеклофенак) с временным эффектом. В 2015 году был установлен диагноз болезни Фабри (снижение активности a-галактозидазы А и мутация с.804А>С гена GLA). При опросе отмечалась четкая связь болей с повышением температуры тела или окружающей среды, в частности очередной приступ развился сразу после поступления в клинику, так как пациент был госпитализирован в палату, находившуюся на "солнечной" стороне отделения. Кроме того, обращало на себя внимание наличие пониженного поотделения и эпизодов диареи с детского возраста. При МРТ признаков поражения сердца и головного мозга не выявлено, в моче белок не определялся, однако при осмотре офтальмологом диагностирована вихревидная кератопатия. В настоящее время пациент получает ферментозаместительную терапию, которая привела к уменьшению частоты приступов и интенсивности нейропатической боли.
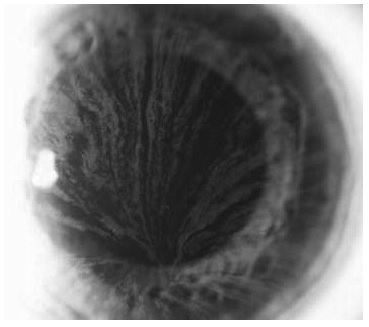
Гипогидроз или ангидроз, связанные с отложением гликосфинголипидов в потовых железах, часто встречаются у пациентов с болезнью Фабри. Нарушение потоотделения обычно появляется уже в детском или подростковом возрасте и объясняет плохую переносимость жары, характерную для этого заболевания. Необходимо отметить, что пациенты обычно сами не жалуются на сниженную потливость, поэтому этот важный симптом может быть упущен при недостаточно тщательном опросе. Еще один типичный симптом болезни Фабри – воронковидная (или вихревидная) кератопатия (cornea verticillata) – коричнево-золотистые отложения в роговице в виде изогнутых линий при отсутствии других причин сходных изменений, например, приема амиодарона или гидроксихлорохина (рис. 4). Вихревидная кератопатия не приводит к нарушению зрения, однако имеет важное значение для подтверждения диагноза болезни Фабри, особенно в сомнительных случаях.
В представленном наблюдении очевидной причиной неправильного диагноза была ошибочная трактовка болей в кистях и стопах, которые отражали наличие нейропатии, а не поражения суставов. Однако, как указано выше, у пациентов с болезнью Фабри могут наблюдаться не только нейропатия, но и боли в суставах и лихорадка, сопровождающиеся лабораторными признаками воспаления. По мнению некоторых экспертов, болезнь Фабри следует включать в алгоритм обследования пациентов с лихорадкой неясного генеза [12]. Среди обследованных нами пациентов боли в суставах отмечались у 6 (7,3%) пациентов, а лихорадка – у 13 (15,9%). Примером может служить следующее наблюдение.
В августе 2015 года в клинику был госпитализирован 30-летний мужчина, которого с 10-летнего возраста беспокоили слабость, быстрая утомляемость, снижение потоотделения и высыпания бурого цвета в области пупка, в последующем распространившиеся на кожу живота и конечностей. В 12 лет появились боли в крупных и мелких суставах конечностей без их припухания и миалгии, которые иногда сопровождались повышением температуры тела до 38-39оС. Чаще всего боли возникали после инсоляции или на фоне повышения температуры тела. Ревматоидный фактор не определялся. Уровень С-реактивного белка был нормальным. Принимал нестероидные противовоспалительные препараты с умеренным эффектом. Предпринимались попытки лечения глюкокортикостероидами, которые вызывали усиление усиление болей. Неоднократно консультировался гематологом, ревматологом, инфекционистом, однако диагноз оставался неясным. С 20 лет присоединились онемение конечностей, чувство их похолодания, парестезии. В июне 2015 года впервые высказано предположение о болезни Фабри, диагноз которой был подтвержден на основании снижения активности a-галактозиды А и результатов молекулярно-генетического исследования (мутация с.55ОТ>G гена GLA). При обследовании в клинике определялись поражение почек (небольшая протеинурия без нарушения функции почек) и вихревидная кератопатия.
В представленном случае доводами против диагноза ревматоидного артрита служили отсутствие припухания и утренней скованности в суставах, ревматоидного фактора и лабораторных признаков воспаления, хотя наличие последних не исключает диагноз болезни Фабри. Мы наблюдали пациентов с подтвержденным диагнозом этого заболевания, у которых помимо типичных симптомов наблюдались частые эпизоды высокой лихорадки, сопровождавшейся повышением СОЭ до 50-80 мм/ч и резким увеличением концентрации С-реактивного белка. E. Verrechia и соавт. в ретроспективном исследовании выявили лихорадку в начале болезни у 12 (20,7%) из 58 пациентов с болезнью Фабри. У 10 из 12 больных имелись акропарестезии, а у 2 были повышены лабораторные маркеры воспаления [13].
Ключом к диагнозу болезни Фабри, как и многих других наследственных заболеваний, может служить не только появление первых симптомов в детском возрасте, но и наличие сходных проявлений у родственников. Так, у 28-летнего брата описанного выше пациента также имелись немногочисленные ангиокератомы, воронковидная кератопатия и микроальбуминурия, а при семейном скрининге мутация с.55ОТ>G гена GLA была выявлена у брата, матери, бабушки по материнской линии, дяди и двоюродных сестер. При анализе семейного анамнеза следует учитывать вариабельность фенотипа болезни Фабри и обращать внимание на наличие не только одинаковых, но и других проявлений этого заболевания (например, терминальная хроническая почечная недостаточность, гипертрофическая кардиомиопатия, инсульт в молодом возрасте и т.п.).
При планировании семейного скрининга необходимо учитывать тип наследования болезни Фабри, которое сцеплено с Х-хромосомой. Мужчина может получить мутантный ген только от матери, в то время как женщине Х-хромосома, содержащая мутантный ген, может быть передана как от отца, так и матери. Соответственно, если заболевание выявлено у мужчины то необходимо обследовать всех его родственников по материнской линии. У женщин, в отличие от мужчин, мутация гена GLA фактически всегда находится в гетерозиготном состоянии, поэтому симптомы болезни Фабри появляются позднее и в целом менее выражены, чем у мужчин, а иногда отсутствуют. Однако женщин не следует считать только носителями мутантного гена, так как у значительной части из них наблюдаются типичные симптомы и развивается поражение внутренних органов, характерное для болезни Фабри.
Поражение периферической нервной системы нередко наблюдается при ревматических болезнях, в том числе ревматоидном артрите, системной красной волчанке, АНЦА-ассоциированных васкулитах. В большинстве случаев оно характеризуется развитием сенсорной полинейропатии, проявляющейся онемением и парестезиями в конечностях. Реже встречаются двигательные расстройства вплоть до тетрапареза, которые при болезни Фабри отсутствуют. Поражение периферической нервной системы при ревматических болезнях не всегда сопровождается болевым синдромом, который, наоборот, типичен для болезни Фабри. Интенсивная боль в конечностях может отмечаться при узелковом полиартериите, однако при этом заболевании неврит практически всегда вызывает моторную дисфункцию, а боль не зависит от температуры окружающей среды, сохраняется постоянно и уменьшается под влиянием иммуносупрессивной терапии.
Поражение почек было выявлено у 70 (85%) из 82 обследованных нами пациентов с болезнью Фабри. Нефропатия проявлялась альбуминурией или небольшой протеинурией, чаще всего не достигавшей нефротического уровня, и прогрессирующим ухудшением функции почек. Артериальная гипертония обычно развивалась только на стадии тяжелой почечной недостаточности. У некоторых пациентов поражение почек было впервые выявлено уже на терминальной стадии хронической почечной недостаточности. Снижение скорости клубочковой фильтрации у пациентов с болезнью Фабри может имитировать картину обострения гломерулонефрита в рамках системного васкулита, что демонстрирует следующее наблюдение.
33-летний мужчина был впервые госпитализирован в клинику в июле 2011 года. С 10-летнего возраста беспокоили боли в кистях и стопах, позднее появились боли в коленных суставах и мышцах. В возрасте 15 лет впервые выявлены небольшая протеинурия и лейкоцитурия. Состояние ухудшилось в возрасте 28 лет, когда было отмечено нарастание протеинурии, повышение сывороточного уровня креатинина и АД. Биопсия почек не проводилась. По поводу предполагаемого обострения гломерулонефрита получал иммуносупрессивную терапию преднизолоном и циклофосфамидом, однако функция почек продолжала ухудшаться. Нарастала артериальная гипертония, которая плохо контролировалась несколькими антигипертензивными препаратами. Обсуждался диагноз системного васкулита, в связи с чем была выполнена биопсия кожно-мышечного лоскута. Признаков васкулита не выявлено. Антитела к цитоплазме нейтрофилов в крови также отсутствовали. В возрасте 30 лет начато лечение программным гемодиализом, а через 2 года произведена трансплантация почки от матери. Учитывая наличие нейропатической боли с детского возраста, множественных ангиокератом в паховой области и характерных черт лица (выступающие супраорбитальные дуги и нижняя челюсть), обсуждался диагноз болезни Фабри, который был подтвержден при молекулярно-генетическом исследовании. С 2014 года получает ферментозаместительную терапию.
Таким образом, как и в предыдущих наблюдениях, заподозрить диагноз болезни Фабри у пациента позволили нейропатическая боль, сохранявшаяся на протяжении более 20 лет, и ангиокератомы. Поражение почек характерно для системных васкулитов с вовлечением мелких сосудов, прежде всего АНЦА-ассоциированных васкулитов – гранулематоза с полиангиитом и микроскопического полиангиита. Без иммуносупрессивной терапии АНЦА-ассоциированный гломерулонефрит быстро прогрессирует и приводит к развитию терминальной хронической почечной недостаточности. Соответственно, появление протеинурии за 13 лет до ухудшения функции почек в описанном случае явно противоречило диагнозу системного васкулита.
У небольшой части пациентов с болезнью Фабри наблюдается "изолированное" поражение почек, не сопровождающееся "классическими" симптомами. В таких случаях диагноз может быть установлен только при электронной микроскопии почечного биоптата или путем скрининга. В 2014-2017 гг. в российских диализных отделениях более чем у 5500 пациентов, получавших лечение программным гемодиализом, была определена активность a-галактозидазы А для исключения болезни Фабри [14]. В результате скрининга диагноз болезни Фабри, подтвержденный при молекулярно-генетическом исследовании, был установлен у 20 пациентов. Следует отметить, что у 16 (80%) из них имелись типичные проявления болезни Фабри, в том числе нейропатическая боль у 16, ангиокератомы у 7 и гипогидроз/ангидроз у 16, которые отмечались с детского возраста. Тем не менее, этот диагноз никогда не обсуждался.
К характерным проявлениям болезни Фабри относятся ишемический инсульт и транзиторные ишемические атаки, развивающиеся в молодом возрасте. Среди 2446 пациентов c этим заболеванием, включенных в Fabry Registry, частота инсульта составила 6,9% у мужчин и 4,3% у женщин (медиана возраста 39,0 и 45,7 лет, соответственно) [15]. Заболеваемость инсультом у пациентов с болезнью Фабри была значительно выше, чем в общей популяции. У большинства больных инсульт развился в возрасте от 20 до 50 лет, в том числе у 30 (21%) из 138 больных – в возрасте до 30 лет. В нашей когорте развитие инсульта было отмечено у 15 (18,3%) из 82 пациентов с болезнью Фабри. В некоторых случаях нарушение мозгового кровообращения было первым проявлением заболевания. В настоящее время мы наблюдаем двух сестер в возрасте 28 и 25 лет, которые перенесли нарушение мозгового кровообращения в возрасте 19 и 23 лет, соответственно. У обеих женщин другие симптомы болезни Фабри до инсульта отсутствовали, а диагноз был установлен около года назад в результате семейного скрининга, когда у сына одной пациентки появились ангиокератомы и были выявлены снижение активности a-галактозидазы А и мутация с.782G>T гена GLA.
Возможными причинами инсульта в молодом возрасте могут быть антифосфолипидный синдром, например, при системной красной волчанке, или васкулит с поражением крупных артерий, например, артериит Такаясу, поэтому пациентов с ранним инсультом обычно направляют на консультацию к ревматологу. В представленных двух наблюдениях заподозрить болезнь Фабри было сложно, хотя развитие инсульта в молодом возрасте у родных сестер позволяло думать о наследственном заболевании.
Диагноз болезни Фабри
Обсуждать диагноз болезни Фабри у взрослого пациента необходимо в следующих случаях:- небольшая протеинурия и нарастающее ухудшение функции почек;
- гипертрофия миокарда неясного происхождения;
- инсульт или транзиторная ишемическая атака в молодом возрасте;
- начало заболевания в детском или подростковом возрасте;
- ранние симптомы (нейропатическая боль, эпизоды болей в суставах, в том числе сопровождающихся лихорадкой, ангиокератомы, воронковидная кератопатия);
- системность поражения (почки, сердце, нервная система, кожа и др.);
- наличие заболевания у родственников.
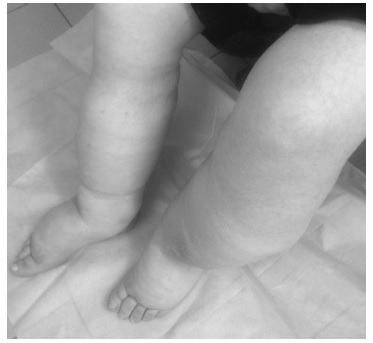
При болезни Фабри могут наблюдаться и другие более редкие проявления. Например, мы наблюдали 48летнего мужчину с подтвержденным диагнозом, у которого с 19 лет помимо нарушений потоотделения отмечалась тяжелая лимфедема нижних конечностей (рис. 5).
Для подтверждения диагноза болезни Фабри определяют активность a-галактозидазы А в плазме или лейкоцитах крови с помощью масс-спектрометрии. Альтернативой может быть исследование высушенных капель крови на специальных бланках, которые могут храниться в течение длительного времени, поэтому их можно отправить в лабораторию по почте. Диагноз подтверждают путем генотипирования гена a-галактозидазы А. У мужчин сниженная активность a-галактозидазы А является достаточно информативным признаком болезни Фабри и может использоваться в качестве скринингового теста. Однако у трети женщин с болезнью Фабри активность этого фермента остается нормальной, поэтому надежно исключить или подтвер дить диагноз позволяет только генетический тест [16]. В последние годы с диагностической целью определяют также уровень глоботриаозилсфингозина (лизо-Gl3). В Москве все необходимые исследования могут быть выполнены в лабораториях Национального медицинского исследовательского центра здоровья детей или Медико-генетического научного центра.
Заключение
Болезнь Фабри – это редкое наследственное заболевание, которое часто остается недиагностированным вследствие низкой осведомленности врачей. Некоторые проявления болезни, в том числе кожные высыпания, нейропатия, системность поражения внутренних органов, могут имитировать ревматические заболевания. Важное значение для диагностики болезни Фабри имеют тщательный опрос пациента, в том числе анализ семейного анамнеза, позволяющий заподозрить наличие наследственного заболевания.
Используемые источники
- Watts RA, Mahr A, Mohammad AJ, et al. Classification, epidemiology and clinical subgrouping of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis. Nephrol Dial Transplant 2015;30 Suppl 1:i14-22.
- Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 2017;91(2):284293.
- Reisin R, Perrin A, Garc ía-Pav ía P. Time delays in the diagnosis and treatment of Fabry disease. Int J Clin Pract. 2017 Jan;71(1). doi: 10.1111/ijcp.12914.
- Моисеев С.В., Новиков П.И., Фомин В.В. Лечение болезни Фабри. Клин фармакол тер 2016;25(4):63-70.
- Моисеев С.В., Новиков П.И., Бровко М.Ю. и др. Отдаленные результаты ферментозаместительной терапии при болезни Фабри. Клин фармакол тер 2015;24(3):34-40.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999;97(1):133-44.
- Рамеев В.В., Козловская Л.В. Аутовоспалительные заболевания: общее понятие, механизмы развития, клиническая картина, подходы к лечению. Нефрология 2012;16(2):49-63.
- De Francesco PN, Mucci J M, Ceci R, et al. Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide. Mol Genet Metab 2013;109:93–9.
- Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab 2017;122(3):19-27.
- Chen KH, Chien Y, Wang KL, et al. Evaluation of proinflammatory prognostic biomarkers for Fabry cardiomyopathy with enzyme replacement therapy. Can J Cardiol 2016;32:1221.e1-1221.e9
- Моисеев С.В., Мершина Е.А., В.Е. Синицын и др. Магнитно-резонансная томография в диагностике поражения сердца при болезни Фабри. Клин фармакол тер 2017;26(3):13-20.
- Manna R, Cauda R, Feriozzi S, et al. Recommendations for the inclusion of Fabry disease as a rare febrile condition in existing algorithms for fever of unknown origin. Intern Emerg Med 2017;12(7):1059-67.
- Verrecchia E, Zampetti A, Antuzzi D, et al. The impact of fever/hyperthermia in the diagnosis of Fabry: A retrospective analysis. Eur J Intern Med 2016;32:26-30.
- Моисеев С.В., Намазова-Баранова Л.С., Савостьянов К.В. и др. Распро -стра ненность и клинические проявления болезни Фабри у диализных пациентов. Клин фармакол тер 2017;26(2):27-33.
- Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke 2009;40(3):788-94.
- Linthorst G, Vedder A, Aerts J, Hollak C. Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers. Clin Chim Acta 2005;353:201-3.