Диагностика и лечение системной склеродермии
Системная склеродермия (ССД) – аутоиммунное заболевание, характеризующееся вовлечением в воспалительный процесс соединительной ткани и проявляющееся поражением кожи, внутренних органов, сосудов и суставов. В статье рассматриваются современные подходы к лечению ССД, которые изложены в недавно опубликованных рекомендациях Европейской антиревматической лиги (EULAR).
Системная склеродермия (ССД) – это аутоиммунное заболевание, в основе которого лежат генерализованная микроангиопатия и активация процессов фиброза кожи и внутренних органов. На ранних стадиях заболевание проявляется кожными изменениями в виде плотного отека пальцев и синдромом Рейно, которые могут не сопровождаться ухудшением общего состояния или признаками поражения внутренних органов (дисфагией, одышкой и др.), поэтому пациенты зачастую не сразу обращаются за медицинской помощью. В связи с этим ССД нередко диагностируют поздно, когда патологические изменения в органах необратимы, а лечение менее эффективно. По данным канадского регистра, у 408 пациентов диагноз ССД был установлен в среднем через 6,0 лет после развития феномена Рейно и через 2,7 года после появления первых “внекожных” проявлений [1]. В России ССД диагностировали через 2,0–2,7 года после появления феномена Рейно при диффузной форме заболевания и через 4,8–6,5 года при лимитированной форме, что связано с разной частой поражения внутренних органов, а также скоростью прогрессирования заболевания [2]. При этом результаты крупного исследования (n=5860) показали, что смертность пациентов с ССД достигает 68 на 1000 человек в год [3]. Таким образом, своевременная диагностика ССД представляет собой сложную, но очень важную задачу для врача.
В настоящее время ССД активно изучается в рамках проекта EUSTAR (European League Against Rheumatism Scleroderma Trials and Research Group). В Российской Федерации в нем принимают участие два центра – НИИ ревматологии им. В.А. Насоновой и клиника им. Е.М. Тареева.
Эпидемиология и факторы риска
Системной склеродермией женщины страдают чаще мужчин (3:1); большая часть пациентов находится в возрасте от 25 до 50 лет [4]. Заболеваемость отличается в разных регионах. Так, в Северной Европе и Японии она составляет менее 10 на 1 млн населения в год, а в Южной Европе, Северной Америке и Австралии достигает 14-21 на 1 млн в год [5]. Распространенность заболевания среди афроамериканцев, американских индейцев, австралийцев, японцев выше, чем среди европейцев и белого населения США [6].
Выявлено множество генов, участвующих в регуляции иммунной системы и повышающих риск развития ССД, в том числе BANK1, C8orf13-BLK, IL-23R, IRF5, STAT4, TBX21 и TNFSF4 [7]. Изучаются также потенциальные эпигенетические механизмы и роль факторов окружающей среды, в том числе кремниевой пыли, органических растворителей, лекарственных препаратов (блеомицина, карбидопы и др.), пестицидов, рапсового масла, кокаина [8].
Клиническая картина
Выделяют две основные формы ССД – диффузную и лимитированную. При лимитированной форме уплотнение кожи отмечается дистальнее локтевых и коленных суставов, при диффузной – изменения кожи могут быть выявлены на туловище, бедрах и плечах (поражение кожи лица встречается при обеих формах). Различия между двумя формами заболевания не ограничиваются распространенностью кожного процесса: диффузная форма характеризуется частым поражением внутренних органов и более быстрым прогрессированием заболевания. Если при диффузной форме ССД 10-летняя выживаемость составляет 65%, то при лимитированной она достигает 92% [9].
Феномен Рейно. Феномен Рейно встречается у 95% больных системной склеродермией и, как правило, оказывается первым признаком болезни [10]. Клинически он имеет две или иногда три стадии – побеление, цианоз и покраснение кожи пальцев, которые развиваются под действием холода и могут сопровождаться онемением и болью [11-15]. Для первичного синдрома Рейно, в отличие от феномена Рейно при ССД, характерны отсутствие изменений при видеокапилляроскопии ногтевого ложа, антинуклеарных антител, симптомов ишемического повреждения тканей (гангрена, язвочки, рубчики), нормальное значение СОЭ [16].
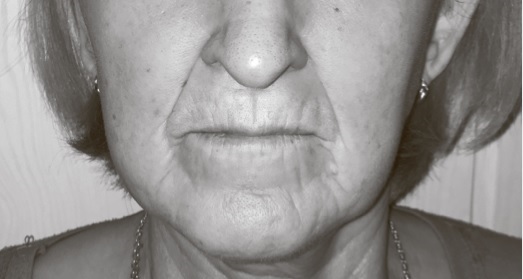
Поражение кожи. Другим признаком ССД является поражение кожи, которое развивается в три стадии: отек (например, плотный отек кистей), уплотнение ( например, склеродактилия), атрофия. На первом этапе отмечаются плотный отек и снижение эластичности кожи и подлежащих тканей, в дальнейшем формируется “склеродерма”, а на стадии атрофии кожа истончается и приобретает цианотично-бурый цвет, появляется специфический блеск, исчезает волосяной покров [17]. Характерны симптом “кисета” (радиальные складки вокруг рта) (рис. 1) и увеличение количества телеангиоэктазий [18]. В результате поражения сосудов микроциркуляторного русла нередко наблюдается ишемическое повреждение кожи – рубчики на дистальных фалангах пальцев по типу “крысиных укусов”, реже – сухие некрозы или гангрена [19]. Существуют и другие специфичные для ССД типы поражения кожи, такие как гипо- и гиперпигментация (“salt and pepper”), кожный кальциноз [20].
Для оценки выраженности кожных изменений применяют модифицированный кожный счет по Rodnan, предполагающий определение толщины кожного покрова в 17 участках тела в баллах от 0 до 3 (нормальная толщина, незначительное, умеренное и выраженное утолщение) с расчетом суммарного балла (максимальное значение 51) [21]. Другим методом, используемым для анализа степени повреждения кожи, является ультразвуковое исследование: гиперэхогенность расценивается как уплотнение кожи вследствие избыточного отложения коллагена, а гипоэхогенность – как признак отека тканей [22]. Реже в клинической практике используют дюрометрию – измерение твердости кожи [23].
Поражение внутренних органов. В России очень большой вклад в изучение висцеральных проявлений ССД внесла проф. Н.Г. Гусева, которая работала в клинике, возглавляемой Е.М. Тареевым, а затем в НИИ ревматологии под руководством акад. В.А. Насоновой [24,25].
У большинства пациентов с ССД (70-98%) развивается поражение желудочно-кишечного тракта, в частности гипотония пищевода, которая проявляется дисфагией и гастро-эзофагеальным рефлюксом. Возможно развитие синдрома мальабсорбции и избыточного роста патогенной флоры на фоне замедления пассажа химуса, а также поражение толстой кишки (диарея, недержание кала) [26]. В отечественном исследовании установлена взаимосвязь между гастроэзофагеальным рефлюксом и тяжестью легочного фиброза [27].
Поражение сердца встречается в 15-35% случаев [28,29] и проявляется сердечной недостаточностью, нарушениями ритма, болевым синдромом [30]. В редких случаях развиваются пороки клапанов, в том числе митральный стеноз [31].
Примерно у 75% больных уже в первые годы заболевания выявляют интерстициальное поражение легких, характеризующееся медленным прогрессирующим течением с исходом в фиброз разной степени тяжести [32]. Л.В. Теплова и соавт. с помощью компьютерной томографии высокого разрешения выявили признаки поражения интерстиция легких у 82% из 138 пациентов с ССД [33].
Для ССД характерно развитие легочной артериальной гипертензии (ЛАГ), иногда тяжелой. По данным недавно опубликованного исследования, в течение в среднем 4 лет умерли 60 из 132 пациентов с ССД, осложнившейся ЛАГ. Выживаемость от момента установления диагноза ЛАГ составила всего 4 (2,2-6,2) года [34]. Легочная гипертензия у пациентов с ССД может быть следствием следующих состояний: ЛАГ (в том числе как исход отложения коллагена в стенке сосудов), легочной веноокклюзионной болезни и/или легочного капиллярного гемангиоматоза, дисфункции левого желудочка, поражения легких с гипоксемией, хронической тромбоэмболической ЛГ [35,36].
Поражение почек встречается у 19% пациентов [29]. Частота развития острого склеродермического криза, проявляющегося быстрым ухудшением функции почек (острое почечное повреждение), составляет 10-15% при диффузной форме ССД и 1-2% при лимитированной форме [37,38]. В настоящее время поражение почек не является основной причиной гибели больных, уступив легочному фиброзу. Склеродермический почечный криз может быть заподозрен при впервые выявленном повышении АД >150/85 мм рт. ст., сохраняющемся в течение последующих 24 ч, а также снижении скорости клубочковой фильтрации на 10% или измеренной СКФ менее 90 мл/мин. Дополнительными признаками склеродермического почечного криза могут быть впервые возникшая гематурия и протеинурия, внезапный отек легких, олигурия или анурия, ретинопатия [39].
Диагностика
ССД следует подозревать у всех пациентов с феноменом Рейно. Важное диагностическое значение имеет поражение кожи (уплотнение кожи, “кисетный” рот, маскообразное лицо, склеродактилия, кальциноз, пигментация), хотя оно может и отсутствовать. При опросе следует также обращать внимание на симптомы поражения внутренних органов, такие как одышку и дисфагию. Для диагностики ССД на практике используют классификационные критерии, разработанные экспертами Американской коллегии ревматологии (ACR) и Европейской антиревматической лиги (EULAR) в 2013 г. (табл. 1) [40]. Необходимо учитывать, что критерии ACR-EULAR мало информативны на ранней и очень ранней стадии ССД, в то время как результаты исследования EUSTAR показали, что срок между развитием синдрома Рейно и других симптомов ССД составляет в среднем 4,8 года при лимитированной форме и 1,9 года при диффузной форме [41]. Полагают, что лечение, начатое в этот период (так называемое “window of opportunity”, или “окно возможностей”), может предотвратить развитие поражения внутренних органов и замедлить прогрессирование заболевания. В связи с этим были разработаны критерии ранней диагностики системной склеродермии (VEDOSS; табл. 2) [42]. На первом этапе диагностики предлагается выявлять ключевые признаки заболевания (так называемые “красные флаги”), такие как синдром Рейно и плотный отек пальцев кистей. На втором этапе проводят видеокапилляроскопию ногтевого ложа и определяют специфические антитела (например, антицентромерные или к топоизомеразе-1) [43]. При очень ранней ССД поражение внутренних органов отсутствует, в то время как при ранней ССД отмечают признаки субклинического их поражения, например, диастолическую дисфункцию левого желудочка по данным эхокардиографии (уменьшение отношения E/A), начальное снижение диффузионной способности легких <80% от должной и/или уменьшение давления в нижнем пищеводном сфинктере <15 мм рт. ст. по данным манометрии [44].
| Критерии | Баллы | |
|---|---|---|
| Утолщение кожи обеих рук выше пястно-фаланговых суставов (достаточный критерий) | 9 | |
| Утолщение кожи пальцев (только больший счет) | ||
| Плотный отек пальцев | 2 | |
| Склеродактилия всех пальцев (дистальнее пястнофаланговых суставов и выше проксимальных межфаланговых суставов) | 4 | |
| Дигитальная ишемия (только больший счет) | ||
| Язвочки | 2 | |
| Рубчики | 3 | |
| Телеангиоэктазии | 2 | |
| Изменения на видеокапилляроскопии | 2 | |
| ЛАГ и/или интерстициальное поражение легких | 2 | |
| Феномен Рейно | 3 | |
| Специфические антитела (ACA, anti-Scl-70, anti-RNA pol III) | 3 | |
| Если сумма баллов составляет 9 и выше, диагностируется систем ная склеродермия | ||
Не стоит забывать о существовании ССД без склеродермы, при которой поражение кожи (уплотнение и фиброз) отсутствует как на ранних, так и поздних стадиях заболевания. В этом случае диагноз устанавливают на основании наличия синдрома Рейно, дигитальных язв, специфических антител, изменений при видеокапилляроскопии, висцеральных поражений [45,46]. Также выделяют CREST-синдром – сочетание кальциноза кожи, синдрома Рейно, нарушения моторики пищевода, склеродактилии и телеангиоэктазий, а также антител к центромере. Для СREST-синдрома характерно достаточно благоприятное течение [47].
| I. Выявление “красных флагов” (феномен Рейно, плотный отек пальцев кистей/склеродактилия) |
| II.а) Проведение видеокапилляроскопии ногтевого ложа (ранние, активные, поздние изменения) |
| б) Серологические исследования: выявление специфических аутоантител (anti-Scl70, ACA, anti-RNA pol III и др.) |
| III. Установление диагноза очень ранней системной склеродермии при выявлении изменений, соответствующих пунктам I и II. |
| IV. Дальнейшее обследование пациентов, соответствующих критериям очень ранней системной склеродермии (компьютерная томография органов грудной клетки, эхокардиография, исследование функции внешнего дыхания, манометрия пищевода). |
| V. Установление диагноза ранней системной склеродермии при выявлении изменений, соответствующих пунктам I, II, IV. |
Важным лабораторным критерием диагностики ССД является наличие аутоантител, например, антител к топоизомеразе I (anti-Scl-70), антицентромерных антител (ACA), антител к рибонуклеопротеазе III (anti-RNA pol III), anti-Th/To, anti-U3RNP/Fibrillarin, anti-Pm/Scl, anti-Ku, антител к белкам сплайсосомы (anti-U1RNP, anti-Sm). В отечественном исследовании (n=300) было показано, что у большинства пациентов с ССД определялись антинуклеарный фактор (83,8%) и anti-Scl-70 (50,0%), реже встречались ACA (14,6%), anti-U1RNP (8,6%) и anti-RNA pol III (5,5%) [48]. Различные типы аутоантител могут ассоциироваться с определенными клиническими проявлениями ССД (табл. 3), поэтому их исследование имеет не только диагностическое, но и прогностическое значение.
| Аутоантитела | Аутоантиген | Клинико-лабораторные ассоциации |
|---|---|---|
| ACA | Центромерный белок |
|
| anti-Scl-70 | ДНК топоизмераза I |
|
| anti-RNA pol III | Мультибелковый комплекс РНК полимеразы III |
|
| anti-Th/To | Мелкие ядерные нуклеопротеины, РНКаза Р и миелоид-связанного белка (mRP) |
|
| anti-U3RNP/Fibrillarin | Комплекс компонентов U3-RNP (U3-RNA,фибрилларин и др.) |
|
| anti-U1RNP | Комплекс компонентов (U1-RNP, А, С, B/В, D-G) |
|
| anti-PM/Scl | Экзосомный белковый комплекс (Pm-Scl-100,Pm-Scl-75) |
|
Важное диагностическое значение имеют изменения при видеокапилляроскопии. Условно выделяют следующие стадии изменений, выявляемых при видеокапилляроскопии ногтевого ложа у больных с ССД: раннюю, активную и позднюю. При склеродермическом типе изменений на разных стадиях могут быть обнаружены гигантские капилляры, капиллярные кровоизлияния, уменьшение количества капилляров или аваскулярные участки, дезорганизация архитектуры капиллярного русла, “ветвистые” капилляры [49].
Лечение
Международные рекомендации по лечению системной склеродермии были разработаны рабочей группой EULAR/EUSTAR в 2016 году [85]. Они подразделены в зависимости от проявлений заболевания (феномен Рейно, язвы дистальных фаланг пальцев, легочная гипертензия, поражение кожи и легких, склеродермический почечный криз, поражение желудочно-кишечного тракта).
Феномен Рейно. Пациентам с феноменом Рейно следует избегать нахождения на холоде и бросить курить. Препаратами первой линии считают дигидропиридиновые антагонисты кальция, например, пролонгиро ванные формы нифедипина или амлодипин. Благоприятное влияние препаратов этой группы на частоту и тяжесть эпизодов ишемии подтверждается результатами мета-анализа клинических исследований [50]. При недостаточной эффективности антагонистов кальция возможно применение ингибиторов фосфоди эстеразы 5 типа (например, силденафила), эффективность которых также была показана при мета-анализе клинических исследований. Однако они несколько чаще вызывают нежелательные явления, в том числе вазомоторные реакции, миалгии, боль в груди, диспепсию и нарушения зрения [51]. При тяжелом феномене Рейно возможно внутривенное введение синтетического аналога простациклина илопроста (0,5-3 нг/кг/мин в течение 3-5 дней) или ингаляционное его применение в дозе 50-150 мкг два раза в сутки. В Российской Федерации для лечения синдрома Рейно чаще при меняют синтетический аналог простагландина Е1 альпростадил. В неконтролируемых клинических исследованиях альпростадил вызывал уменьшение или купирование боли, ускорял заживление язв и улучшал показатели микроциркуляции [24,52]. В 1985 году было проведено многоцентровое, двойное слепое, плацебоконтролируемое исследование, в которое было включено 55 пациентов с первичным феноменом Рейно и феноменом Рейно в рамках ССД. 72-часовое внутривенное введение альпростадила улучшало кровоток в микроциркуляторном русле по сравнению с плацебо [53].
При непереносимости вазодилататоров в качестве альтернативного препарата в рекомендациях указан флуоксетин 20 мг/сут (селективный ингибитор обратного захвата серотонина), хотя он изучался только в одном небольшом исследованием (n=27) пациентов), в котором по эффективности превосходил нифедипин у пациентов с тяжелым феноменом Рейно [54].
Обсуждается возможность применения топических аналогов нитроглицерина для лечения феномена Рейно. Так, в многоцентровом, двойном слепом, плацебоконтролируемом исследовании MQX-503 (аналог нитроглицерина, быстро абсорбирующийся с поверхности кожи) улучшал микроциркуляцию у пациентов с феноменом Рейно [55]. Длительное время считалось, что блокаторы эндотелиновых рецепторов (бозентан) эффективны только в профилактике новых дигитальных язв, но мало влияют на течение феномена Рейно. Однако недавно было показано, что бозентан может применяться для лечения феномена Рейно у пациентов с ССД, которым противопоказана терапия простаноидами [56,57].
Дигитальные язвы. Пациентам с дигитальными язвами следует избегать нахождения на холоде и бросить курить. Основными препаратами считают простаноиды (например, внутривенное введения илопроста) и ингибиторы фосфодиэстеразы 5 типа, эффективность которых подтверждена результатами мета-анализов клинических исследований [58]. Из ингибиторов фосфодиэстеразы 5 типа у пациентов с дигитальными язвами лучше всего изучен тадалафил, в то время как результаты применения силденафила оказались достаточно противоречивыми. По данным одного из последних исследований SEDUCE, при его применении частота возникновения язвенных дефектов была даже выше, чем в группе плацебо [59]. Тем не менее, авторы рекомендаций отмечают, что доза силденафила в указанном исследовании была ниже (60 мг/сут), чем в других исследованиях (100 мг/сут). При неэффективности ингибиторов фосфодиэстеразы может быть назначен бозентан (неселективный антагонист ЕТА и ЕТВ эндотелиновых рецепторов), эффективность которого была установлена в двух рандомизированных исследованиях (RAPIDS-1, RAPIDS-2). В этих исследованиях лечение бозентаном вызывало уменьшение количества новых язвенных дефектов, но не способствовало заживлению уже имевшихся язв [56,60]. Бозентан может оказывать гепатотоксическое действие, а также обладает тератогенностью. При этом он снижает эффективность пероральных контрацептивов, ингибируя систему цитохрома Р450. Таким образом, ввиду скорее профилактического действия препарата и серьезности нежелательных явлений, бозентан считается препаратом третьей линии [61]. В острую фазу или при подозрении на тромботические осложнения к терапии могут быть присоединены антиагреганты (аспирин, клопидогрел) и антикоагулянты (эноксапарин, варфарин) [58,62,63]. При наличии некротизированной ткани в области ишемического дефекта необходима первичная хирургическая обработка раны, назначение антибактериальной терапии при инфицировании язвы, применение специальных повязок для улучшения заживления язвы [64-66]. Для купирования болевого синдрома применяют нестероидные противовоспалительные препараты, опиоиды, лидокаин [62].
Легочная артериальная гипертензия. Применение антагонистов эндотелиновых рецепторов, таких как бозентан, амбризентан, мацитентан, приводит к повышению толерантности к физическим нагрузкам и замедляет прогрессирование легочной гипертензии [6769]. Зависимость эффекта от дозы была отмечена только при лечении амбризентаном (доза 10 мг по эффективности превосходила дозу 5 мг). К такому же эффекту приводило применение ингибиторов фосфодиэстеразы 5 типа, в том числе силденафила (от 60 до 240 мг/сут) и тадалафила (40 мг/сут), а также риоцигуата – стимулятора растворимой гуанилатциклазы [67,69,70]. При этом эффективность риоцигуата (улучшение пробы с 6-минутной ходьбой) была доказана у пациентов как с ЛАГ, так и с хронической тромбоэмболической легочной гипертензией в исследованиях PATENT и CHEST [71]. Более высокая эффективность комбинированной терапии бозентаном и силденафилом у пациентов с ЛАГ (в том числе идиопатической, наследственной и в рамках диффузных заболеваний соединительной ткани) не была подтверждена в исследовании COMPASS [72]. При тяжелой ЛАГ возможно внутривенное применение эпопростенола (стартовая доза 2 нг/кг/мин), которое в двух исследованиях приводило к улучшению толерантности к физическим нагрузкам и показателей гемодинамики [73,74]. Недостатком препарата является короткий период полувыведения, в связи чем его приходится вводить с помощью центрального венозного катетера, а также синдром отмены, характеризующийся резким повышением давления в легочной артерии при прекращении лечения. Альтернативой может служить ингаляционное применение илопроста, хотя следует учитывать, что его эффективность в лечении ЛАГ у пациентов с ССД недостаточно изучена [67,69]. Ранее пациентам с ЛАГ назначали терапию варфарином, однако в регистре COMPERA не была подтверждена эффективность антикоагулянтов у пациентов с ССД [75,76].
Поражение кожи. Ранние кожные изменения при ССД могут поддаваться лечению метотрексатом. В двух рандомизированных клинических исследованиях препарат изучался в дозах 15 мг/нед в течение 2 лет и 10 мг/нед в течение года. Эти дозы ниже доз препарата, которые применяются при других ревматических заболеваниях, в частности ревматоидном артрите [40,77]. Применение циклофосфамида у пациентов с висцеральными проявлениями ССД в рандомизированных клинических исследованиях также приводило к улучшению состояния кожи [78]. Эффективность микофенолата мофетила и азатиоприна изучена недостаточно.
Интерстициальная болезнь легких. В рандомизированном клиническом исследовании (Scleroderma Lung Study) лечение циклофосфамидом в дозе 1-2 мг/кг/сут внутрь вызывало уменьшение одышки и изменений на компьютерных томограммах, улучшение качества жизни и показателей вентиляционной функции, причем наиболее выраженный эффект был отмечен у пациентов с тяжелым поражением легких [78]. В другом многоцентровом, проспективном, рандомизированном, двойном слепом, плацебо-контролируемом исследовании циклофосфамид применяли внутривенно в дозе 600 мг/м2/мес в течение 6 месяцев, а затем заменяли на азатиоприн (2,5 мг/кг/сут) в течение 6 месяцев. В этом исследовании был выявлен прирост форсированной жизненной емкости (ФЖЕЛ) легких в основной группе на 4,2% по сравнению с контролем [79]. Был сделан вывод, что циклофосфамид замедляет прогресси ро вание заболевания, а его применение оправдано у пациентов с прогрессирующим интерстициальным поражением легких. Режим дозирования и длительность терапии следует подбирать индивидуально, учитывая эффективность и токсичность препарата. Следует отметить, что при анализе отдаленных результатов исследования (через 24 месяца) было выявлено ухудшение показателей функции внешнего дыхания и рентгенологической картины, которые фактически возвращались к исходным значениям [80]. В рандомизированном двойном слепом исследовании была сопоставлена эффективность микофенолата мофетила (3000 мг/сут в течение 24 мес) и циклофосфамида внутрь (2 мг/кг/сут в течение 12 мес с дальнейшим приемом плацебо в течение 12 мес) у пациентов с ССД. Улучшение показателей функции внешнего дыхания было сопоставимым в двух группах, однако переносимость микофенолата мофетила была лучше, чем циклофосфамида [81].
В исследовании случай-контроль лечение ритуксимабом пациентов с ССД и интерстициальным поражением легких предотвращало снижение ФЖЕЛ по сравнению с контролем (p=0,02) [82]. Л.П. Ананьева и соавт. применяли ритуксимаб у 27 пациентов с ССД и поражением легких. У 82% пациентов был достигнут хороший эффект, а у 15% – удовлетворительный. Через 1 год после первого введения ритуксимаба было выявлено достоверное увеличение среднего значения ФЖЕЛ. Эффективность препарата у пациентов с длительностью болезни менее 5 лет была выше, чем у больных с более длительным анамнезом ССД: увеличение ФЖЕЛ на 11,4±12,2% и 3,7±5,7%, соответственно [83]. Мы применяли ритуксимаб у 8 пациентов с ССД и интерстициальным поражением легких. Через 6 мес ФЖЕЛ и диффузионная способность легких увеличились на 6,9±6,9% и 4,7±4,7% соответственно, а через 12 месяцев – на 8,9±6,9% и 6,7±4,6%.
В рандомизированном, двойном-слепом, плацебоконтролируемом исследовании лечение тоцилизумабом, блокирующим рецепторы интерлейкина-6, у 87 пациентов с ССД и интерстициальным поражением легких задерживало прогрессирование фиброза через 24 и 48 недель [84].
Склеродермический почечный криз. Основа лечения – ингибиторы АПФ, в частности каптоприл, который назначают в дозе 6,25-12,5 мг три раза в сутки, а затем постепенно увеличивают дозу до максимальной (50 мг три раза в сутки) [85]. Каптоприл не отменяют, даже если функция почек продолжает ухудшаться. После стабилизации АД можно перейти на прием ингибитора АПФ длительного действия. Постоянный прием ингибитора АПФ увеличил выживаемость пациентов со склеродермическим почечным кризом с 10% в течение года до 60% в течение 5 лет [86,87]. Если на фоне приема каптоприла в максимальной дозе не удается добиться нормализации АД в течение 72 ч, добавляют блокаторы кальциевых каналов, нитраты (особенно при появлении застойных явлений в легких) или другие вазодилатирующие средства. При сохранении олигурии может потребоваться лечение гемодиализом. Медлен ное (в течение 2 лет) восстановление или улучшение функции почек после склеродермического почечного криза происходит у 30% пациентов [88]. Если через 2 года сохраняется потребность в гемодиализе, возможна трансплантация почки [89].
Интересно отметить, что независимым фактором риска почечного склеродермического криза считают применение глюкокортикостероидов в дозе более 15 мг/сут, что недавно было показано в двух крупных исследованиях [90,91]. Возможно также развитие так называемого “нормотензивного” почечного криза, при котором АД не превышает 140/90 мм рт. ст. Выживаемость пациентов с нормотензивным кризом значительно ниже таковой пациентов с гипертензивным кризом – 13% и 35%, соответственно, ввиду поздней диагностики [92]. Рекомендаций по лечению данного состояния нет, в связи с чем терапия аналогична лечению гипертензивного почечного криза.
Поражение желудочно-кишечного тракта. При наличии гастроэзофагеального рефлюкса рекомендуется применение ингибиторов протонной помпы [93]. При необходимости могут быть использованы прокинетики [94]. Лечение антибактериальными препаратами для профилактики избыточного бактериального роста в тонкой кишке представляется спорным, учитывая отсутствие соответствующих рандомизированных клинических исследований [95].
Ранняя ССД. Количество исследований, в которых изучались подходы к лечению пациентов с ранней ССД, ограничено. В достаточно крупном проспективном обсервационном исследовании ESOS (European Scleroderma Observational Study) у 326 пациентов c диффузной формой ССД длительностью не более 3 лет изучалась эффективность различных режимов иммуносупрессивной терапии, в том числе метотрексатом (n=65), микофенолата мофетилом (n=118) и циклофосфамидом (n=87). Пятьдесят шесть пациентов не получали иммуносупрессивную терапию. 276 (84,7%) пациентов завершили 12-месячное наблюдение и 234 (71,7%) – 24-месячное. Во всех группах пациентов, получавших иммуносупрессивные препараты, отмечалось значительное снижение кожного счета по Rodnan. Через 24 месяца выживаемость была ниже в контрольной группе пациентов, хотя разница между группами не достигла статистической значимости [96].
Применение глюкокортикостероидов. Применение глюкокортикостероидов для лечения ССД по возможности следует ограничивать в связи с риском развития отдаленных осложнений, однако при прогрессирующем диффузном поражении кожи и других клинических признаках воспалительной активности (серозит, миозит, прогрессирующее интерстициальное поражение легких, рефрактерный синовит и/или теносиновит) целесообразно назначение глюкокортикостероидов в дозах до 15 мг/сут, так как прием этих препаратов в более высоких дозах увеличивает риск развития склеродермического почечного криза.
Заключение
ССД остается одним из самых неблагоприятных диффузных заболеваний соединительной ткани ввиду частого тяжелого поражения жизненно важных органов и ограниченных возможностей терапии. Многие вопросы лечения ССД остаются нерешенными, а прогнозирование эффективности терапии представляется затруднительным в связи с малым количеством исследований. Концепция ранней и очень ранней ССД, а также “окна возможностей” представляет интерес, однако опыт назначения иммуносупрессивного лечения в столь ранние сроки весьма ограничен. Перспективно дальнейшее изучение эффективности ритуксимаба, тоцилизумаба и, возможно, других ГИБП, однако до сих пор остается неясным, имеют ли препараты данной группы реальное преимущество перед стандартными препаратами.
Используемые источники
- Hudson M, Thombs B, Baron M, et al. Time to diagnosis in systemic sclerosis: is sex a factor? Arthritis Rheum 2009;61(2):274-8.
- Ананьева Л.П. Новые классификационные критерии системной склеродермии (лекция). Научно-практическая ревматология 2013;51(5):539-44.
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010;69(10):1809-15.
- Silman AJ. Scleroderma demographics and survival. J Rheumatol Suppl 1997;48:58-61.
- Barnes J,Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012;24(2):165-70.
- Ranque B, Mouthon L. Geoepidemiology of systemic sclerosis. Autoimmun Rev 2010;9(5):A311-8.
- Agarwal SK,Reveille JD. The genetics of scleroderma (systemic sclerosis). Curr Opin Rheumatol 2010;22(2):133-8.
- Mora GF. Systemic sclerosis: environmental factors. J Rheumatol 2009;36(11): 2383-96.
- Al-Dhaher FF, Pope JE,Ouimet JM. Determinants of morbidity and mortality of systemic sclerosis in Canada. Semin Arthritis Rheum 2010;39(4):269-77.
- Hinchcliff M,Varga J. Systemic sclerosis/scleroderma: a treatable multisystem disease. Am Fam Physician 2008;78(8):961-8.
- Brown KM, Middaugh SJ, Haythornthwaite JA, et al. The effects of stress, anxiety, and outdoor temperature on the frequency and severity of Raynaud's attacks: the Raynaud's Treatment Study. J Behav Med 2001;24(2):137-53.
- Wigley FM,Flavahan NA. Raynaud's Phenomenon. N Engl J Med 2016;375(6): 556-65.
- Sulli A, Ruaro B, Alessandri E, et al. Correlations between nailfold microangiopathy severity, finger dermal thickness and fingertip blood perfusion in systemic sclerosis patients. Ann Rheum Dis 2014;73(1):247-51.
- Ruaro B, Smith V, Sulli A, et al. Methods for the morphological and functional evaluation of microvascular damage in systemic sclerosis. Korean J Intern Med 2015;30(1):1-5.
- Herrick AL. The pathogenesis, diagnosis and treatment of Raynaud phenomenon. Nat Rev Rheumatol 2012;8(8):469-79.
- LeRoy EC, Medsger TA. Raynaud's phenomenon: a proposal for classification. Clin Exp Rheumatol 1992;10(5):485-8.
- Akesson A, Hesselstrand R, Scheja A, et al. Longitudinal development of skin involvement and reliability of high frequency ultrasound in systemic sclerosis. Ann Rheum Dis 2004;63(7):791-6.
- Derk CT, Huaman G, Littlejohn J, et al. Predictors of early mortality in systemic sclerosis: a case-control study comparing early versus late mortality in systemic sclerosis. Rheumatol Int 2012;32(12):3841-4.
- Hughes M,Herrick AL. Digital ulcers in systemic sclerosis. Rheumatology (Oxford) 2017;56(1):14-25.
- Aringer M, Muller-Ladner U, Burkhardt H, et al. [Common German language nomenclature for systemic sclerosis]. Z Rheumatol 2015;74(2):100-3.
- Clements P, Lachenbruch P, Siebold J, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol 1995;22(7):1281-5.
- Hesselstrand R, Scheja A, Wildt M, et al. High-frequency ultrasound of skin involvement in systemic sclerosis reflects oedema, extension and severity in early disease. Rheumatology (Oxford) 2008;47(1):84-7.
- Kissin EY, Schiller AM, Gelbard RB, et al. Durometry for the assessment of skin disease in systemic sclerosis. Arthritis Rheum 2006;55(4):603-9.
- Гусева Н.Г. Алекперов Р.Т. Вазапростан при системной склеродермии. Клиническая ревматология 1997(4):5-9.
- Тареев Е.М., Гусева Н.Г. Пороки сердца при коллагенозах. Российский медицинский журнал 1967;4:3.
- Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol 2013;19(41):7062-8.
- Сосновская А.В., Фомин В.В., Попова Е.Н., и др. Гастроэзофагеально-рефлюксная болезнь и взаимосвязь с выраженностью поражения легких при системной склеродермии. Клиническая нефрология 2016;1:24-8.
- Steen VD, Medsger TA, Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000;43(11):2437-44.
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002;81(2):139-53.
- Богданов А.П., Моисеев С.В. Поражение сердца при системной склеродермии: клинические аспекты и современные методы диагностики. Терапев ти ческий архив 1994;66(5):87-91.
- Kahan A, Allanore Y. Primary myocardial involvement in systemic sclerosis. Rheumatology (Oxford) 2006;45 Suppl 4:iv14-7.
- Bussone G, Mouthon L. Interstitial lung disease in systemic sclerosis. Autoimmun Rev 2011;10(5):248-55.
- Теплова Л.В., Ананьева Л.П., Лесняк В.Н. и др. Системная склеродермия с интерстициальным поражением легких: сравнительная клиническая характеристика с больными без поражения легких. Научно-практическая ревматология 2010;3:36-41.
- Morrisroe K, Stevens W, Huq M, et al. Survival and quality of life in incident systemic sclerosis-related pulmonary arterial hypertension. Arthritis Res Ther 2017;19(1):122.
- Galie N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016;37(1):67-119.
- Vachiery JL,Coghlan G. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev 2009;18(113):162-9.
- Penn H, Howie AJ, Kingdon EJ, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM 2007;100(8):485-94.
- Teixeira L, Mouthon L, Mahr A, et al. Mortality and risk factors of scleroderma renal crisis: a French retrospective study of 50 patients. Ann Rheum Dis 2008;67(1):110-6.
- Bar J, Ehrenfeld M, Rozenman J, et al. Pulmonary-renal syndrome in systemic sclerosis. Semin Arthritis Rheum 2001;30(6):403-10.
- van den Hoogen FH, Boerbooms AM, Swaak AJ, et al. Comparison of methotrexate with placebo in the treatment of systemic sclerosis: a 24 week randomized double-blind trial, followed by a 24 week observational trial. Br J Rheumatol 1996;35(4):364-72.
- Walker UA, Tyndall A, Czirjak L, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Ann Rheum Dis 2007;66(6):754-63.
- Bellando-Randone S, Matucci-Cerinic M. From Raynaud's phenomenon to very early diagnosis of systemic sclerosis The VEDOSS approach. Curr Rheumatol Rev 2013;9(4):245-8.
- Minier T, Guiducci S, Bellando-Randone S, et al. Preliminary analysis of the very early diagnosis of systemic sclerosis (VEDOSS) EUSTAR multicentre study: evidence for puffy fingers as a pivotal sign for suspicion of systemic sclerosis. Ann Rheum Dis 2014;73(12):2087-93.
- Valentini G, Cuomo G, Abignano G, et al. Early systemic sclerosis: assessment of clinical and pre-clinical organ involvement in patients with different disease features. Rheumatology (Oxford) 2011;50(2):317-23.
- Diab S, Dostrovsky N, Hudson M, et al. Systemic sclerosis sine scleroderma: a multicenter study of 1417 subjects. J Rheumatol 2014;41(11):2179-85.
- Poormoghim H, Lucas M, Fertig N, et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum 2000;43(2):444-51.
- Adigun R,Bhimji SS, Systemic Sclerosis (CREST syndrome), in StatPearls. 2017: Treasure Island (FL).
- Старовойтова М.Н., Десинова О.В., Конева О.А. Профиль аутоантител при системной склеродермии. Научно-практическая ревматология 2016;54(4): 418-23.
- Cutolo M, Herrick AL, Distler O, et al. Nail fold videocapillaroscopic features and other clinical risk factors for digital ulcers in systemic sclerosis: A multicenter, prospective cohort study. Arthritis Rheumatol 2016;68(10):2527-39.
- Thompson AE, Shea B, Welch V, et al. Calcium-channel blockers for Raynaud's phenomenon in systemic sclerosis. Arthritis Rheum 2001;44(8):1841-7.
- Roustit M, Blaise S, Allanore Y, et al. Phosphodiesterase-5 inhibitors for the treatment of secondary Raynaud's phenomenon: systematic review and metaanalysis of randomised trials.Ann Rheum Dis 2013;72(10):1696-9.
- Гусева Н.Г. Вазапростан в комплексном лечении системной склеродермии и синдрома Рейно. Врач 2006(5):46-50.
- Mohrland JS, Porter JM, Smith EA, et al. A multiclinic, placebo-controlled, double-blind study of prostaglandin E1 in Raynaud's syndrome. Ann Rheum Dis 1985;44(11):754-60.
- Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud's phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 2001;40(9):1038-43.
- Hummers LK, Dugowson CE, Dechow FJ, et al. A multi-centre, blinded, randomised, placebo-controlled, laboratory-based study of MQX-503, a novel topical gel formulation of nitroglycerine, in patients with Raynaud phenomenon. Ann Rheum Dis 2013;72(12):1962-7.
- Matucci-Cerinic M, Denton CP, Furst DE, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis 2011;70(1):32-8.
- Parisi S, Bruzzone M, Centanaro Di Vittorio C, et al. Efficacy of bosentan in the treatment of Raynaud's phenomenon in patients with systemic sclerosis never treated with prostanoids. Reumatism 2014;65(6):286-91.
- Tingey T, Shu J, Smuczek J, et al. Meta-analysis of healing and prevention of digital ulcers in systemic sclerosis. Arthritis Care Res (Hoboken) 2013;65(9):1460-71.
- Hachulla E, Hatron PY, Carpentier P, et al. Efficacy of sildenafil on ischaemic digital ulcer healing in systemic sclerosis: the placebo-controlled SEDUCE study. Ann Rheum Dis 2016;75(6):1009-15.
- Korn JH, Mayes M, Matucci Cerinic M, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum 2004;50(12):3985-93.
- Kuwana M, Okazaki Y,Kaburaki J. Long-term beneficial effects of statins on vascular manifestations in patients with systemic sclerosis. Mod Rheumatol 2009;19(5):530-5.
- Cappelli L,Wigley FM. Management of Raynaud phenomenon and digital ulcers in scleroderma. Rheum Dis Clin North Am 2015;41(3):419-38.
- Garcia JG, Pavon BM, Martin LM, et al. Lipschutz ulcer: A cause of misdiagnosis when suspecting child abuse. Am J Emerg Med 2016;34(7):1326 e1-2.
- Amanzi L, Braschi F, Fiori G, et al. Digital ulcers in scleroderma: staging, characteristics and sub-setting through observation of 1614 digital lesions. Rheumatology (Oxford) 2010;49(7):1374-82.
- Fraenkel L. Raynaud's phenomenon: epidemiology and risk factors. Curr Rheumatol Rep 2002;4(2):123-8.
- Khanna D, Denton CP, Merkel PA, et al. Effect of macitentan on the development of new ischemic digital ulcers in patients with systemic sclerosis: DUAL-1 and DUAL-2 randomized clinical trials. JAMA 2016;315(18):1975-88.
- Ghofrani HA, Distler O, Gerhardt F, et al. Treatment of pulmonary arterial hypertension (PAH): updated Recommendations of the Cologne Consensus Conference 2011. Int J Cardiol 2011;154 Suppl 1:S20-33.
- Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 2013;369(9):809-18.
- Taichman DB, Ornelas J, Chung L, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST guideline and expert panel report. Chest 2014;146(2):449-75.
- Ghofrani HA, Galie N, Grimminger F, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369(4):330-40.
- Lian TY, Jiang X,Jing ZC. Riociguat: a soluble guanylate cyclase stimulator for the treatment of pulmonary hypertension. Drug Des Devel Ther 2017;11:1195207.
- McLaughlin V, Channick RN, Ghofrani HA, et al. Bosentan added to sildenafil therapy in patients with pulmonary arterial hypertension. Eur Respir J 2015;46(2): 405-13.
- McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002;106(12):1477-82.
- Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002;40(4):780-8.
- Olsson KM, Delcroix M, Ghofrani HA, et al. Anticoagulation and survival in pulmonary arterial hypertension: results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014;129(1):57-65.
- Preston IR, Roberts KE, Miller DP, et al. Effect of warfarin treatment on survival of patients with pulmonary arterial hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation 2015;132(25):2403-11.
- Pope JE, Bellamy N, Seibold JR, et al. A randomized, controlled trial of methotrexate versus placebo in early diffuse scleroderma. Arthritis Rheum 2001;44(6):1351-8.
- Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med 2006;354(25):2655-66.
- Hoyles RK, Ellis RW, Wellsbury J, et al. A multicenter, prospective, randomized, double-blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum 2006;54(12):3962-70.
- Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med 2007;176(10):1026-34.
- Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med 2016;4(9):708-19.
- Jordan S, Distler JH, Maurer B, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group.Ann Rheum Dis 2015;74(6):1188-94.
- Ананьева Л.П., Десинова О.В., Конева О.А. Лечение ритуксимабом интерстициального поражения легких при системной склеродермии. Научнопрактическая ревматология 2013;51(5):514-23.
- Khanna D, Denton CP, Jahreis A, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet 2016;387(10038):2630-40.
- Kowal-Bielecka O, Landewe R, Avouac J, et al. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR). Ann Rheum Dis 2009;68(5):620-8.
- Steen VD, Costantino JP, Shapiro AP, et al. Outcome of renal crisis in systemic sclerosis: relation to availability of angiotensin converting enzyme (ACE) inhibitors. Ann Intern Med 1990;113(5):352-7.
- Steen VD,Medsger TA, Jr. Long-term outcomes of scleroderma renal crisis. Ann Intern Med 2000;133(8):600-3.
- Steen VD. Scleroderma renal crisis. Rheum Dis Clin North Am 2003;29(2):315-33.
- Федеральные клинические рекомендации по диагностике и лечению системной склеродермии. Москва, 2013.
- Guillevin L, Berezne A, Seror R, et al. Scleroderma renal crisis: a retrospective multicentre study on 91 patients and 427 controls. Rheumatology (Oxford) 2012;51(3):460-7.
- Montanelli G, Beretta L, Santaniello A, et al. Effect of dihydropyridine calcium channel blockers and glucocorticoids on the prevention and development of scleroderma renal crisis in an Italian case series. Clin Exp Rheumatol 2013;31(2 Suppl 76):135-9.
- Helfrich DJ, Banner B, Steen VD, et al. Normotensive renal failure in systemic sclerosis. Arthritis Rheum 1989;32(9):1128-34.
- Pakozdi A, Wilson H, Black CM, et al. Does long term therapy with lansoprazole slow progression of oesophageal involvement in systemic sclerosis? Clin Exp Rheumatol 2009;27(3 Suppl 54):5-8.
- Wang SJ, Lan JL, Lan JL, et al. Effects of cisapride on colonic transit in patients with progressive systemic sclerosis. Clin Rheumatol 2002;21(4):271-4.
- Marie I, Ducrotte P, Denis P, et al. Small intestinal bacterial overgrowth in systemic sclerosis. Rheumatology (Oxford) 2009;48(10):1314-9.
- Herrick AL, Pan X, Peytrignet S, et al. Treatment outcome in early diffuse cutaneous systemic sclerosis: the European Scleroderma Observational Study (ESOS). Ann Rheum Dis 2017;76(7):1207-18