Идиопатический легочный фиброз (ИЛФ): современный подход к классификации и диагностике
Идиопатический легочный фиброз (ИЛФ) – это вариант идиопатической интерстициальной пневмонии (ИИП), характеризующийся неуклонным прогрессирующим течением и высокой смертностью. В отличие от большинства ИИП, иммуносупрессивная терапия не оказывает влияния на скорость прогрессирования ИЛФ. В течение последнего десятилетия установлена эффективность двух антифибротических препаратов в лечении ИЛФ – пирфенидона и нинтеданиба. Чтобы своевременно начать патогенетическую терапию, необходимо как можно быстрее установить диагноз ИЛФ на основании диагностического алгоритма, предполагающего анализ клинических, лабораторных и инструментальных данных, прежде всего результатов компьютерной томографии высокого разрешения (КТВР). При недостаточной информативности последней может быть использована малоинвазивная трансбронхиальная криобиопсия легкого, которая по точности сопоставима с хирургической биопсией легкого. Продолжается поиск молекулярно-биологических и генетических маркеров ИЛФ.
Согласно классификации Американского торакального общества/Европейского респираторного общества (ATS):1–112. /ERS):1–112. ) [1], идиопатический легочный фиброз (ИЛФ) представляет собой форму идиопатической интерстициальной пневмонии (ИИП) (табл. 1). Доля ИЛФ составляет 20-30% в структуре всех ИИП, а заболеваемость – от 7 до 17 случаев на 100 000 населения [2]. Мужчины болеют несколько чаще, чем женщины (соотношение мужчин/женщин приблизительно 1,5:1) [3]. ИЛФ развивается в основном у людей среднего и пожилого возраста: возраст 65% пациентов на момент постановки диагноза составляет 60 лет и более [4].
| Частые формы ИИП |
| Идиопатический легочный фиброз (ИЛФ) |
| Идиопатическая неспецифическая интерстициальная пневмония |
| Респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких |
| Десквамативная интерстициальная пневмония |
| Криптогенная организующая пневмония |
| Острая интерстициальная пневмония |
| Редкие формы ИИП |
| Идиопатическая лимфоцитарная интерстициальная пневмония |
| Идиопатический плевропаренхиматозный фиброэластоз |
| Неклассифицируемые формы (ИИП) |
В 2018 году P. Wolters и соавт. предложили выделять 4 варианта легочного фиброза в зависимости от патогенеза заболевания (табл. 2) [5]. ИЛФ характеризуется прогрессирующим течением с развитием дыхательной недостаточности и среди всех ИИП обладает самым неблагоприятным прогнозом: средняя выживаемость составляет от 2 до 5 лет [6,7]. Высокая смертность пациентов с ИЛФ объясняется особенностями патогенеза заболевания – преобладанием фиброза при незначительной выраженности воспалительных изменений [8,9]. Основным механизмом, приводящим к развитию прогрессирующего легочного фиброза, является персистирующее повреждение альвеолярного эпителия с последующим нарушением процессов его регенерации, избыточным отложением компонентов внеклеточного матрикса, активацией фибробластов и миофибробластов [10]. Указанные изменения определяют неэффективность традиционной иммуносупрессивной терапии у пациентов с ИЛФ [11]. Тем не менее, в настоящее время достигнуты значительные успехи в лечении ИЛФ, связанные с применением антифибротических препаратов – пирфенидона (антагониста трансформирующего фактора роста бета – TGF β) и нинтеданиба (множественного ингибитора тирозинкиназ), замедляющих уменьшение легочных объемов, в первую очередь, форсированной жизненной емкости легких (ФЖЕЛ), и улучшающих выживаемость без прогрессирования заболевания [12]. При отсутствии противопоказаний трансплантация легких также рассматривается в качестве варианта лечения у пациентов с прогрессирующим ИЛФ, осложнившимся тяжелой дыхательной недостаточностью [13,14].
| Группа 1: ЛФ, индуцированный дисфункцией эпителиальных клеток | ИЛФ |
| Группа 2: ЛФ, индуцированный дисфункцией клеток воспалени | Системная склеродермия, ревматоидный артрит, синдром Шегрена, экзогенный аллергический альвеолит, саркоидоз, НСИП |
| Группа 3: ЛФ, вызванный приемом лекарственных препаратов или воздействием профессиональных факторов | Асбестоз, силикоз, лекарственное поражение легких |
| Группа 4: ЛФ, связанный с курением | Десквамативная интерстициальная пневмония, респираторный бронхиолит, ассоциированный с интерстициальным заболеванием легких, Лангерганс-клеточный гистиоцитоз |
Клиническая картина
Основные жалобы у пациентов с ИЛФ – прогрессирующая одышка и сухой кашель, усиливающиеся при физической нагрузке. Реже отмечаются боль и дискомфорт в грудной клетке, повышенная утомляемость, общая слабость, снижение массы тела. В ряде случаев заболевание на начальных этапах протекает бессимптомно, а первыми проявлениями оказываются изменения функциональных легочных параметров [1]. Типичным аускультативным феноменом при ИЛФ является крепитация, преимущественно в задне-базальных отделах легких. У больных c развернутой стадией ИЛФ могут отмечаться признаки вторичной артериальной легочной гипертензии с развитием легочного сердца и правожелудочковой сердечной недостаточности [15].
При ИЛФ может определяться незначительное повышение СОЭ. Несмотря на наличие прогрессирующей дыхательной недостаточности, выраженное увеличение концентрации гемоглобина наблюдается крайне редко. уменьшением всех легочных объемов в сочетании со снижением диффузионной способности легких (DLCO). Одним из ранних проявлений ИЛФ может быть изолированное снижение DLCO при относительной сохранности легочных объемов. Также к ранним проявлениям ИЛФ относят увеличение альвеолоартериального градиента по кислороду, что часто характеризуется нормальными показателями сатурации крови в покое и десатурацией при физической нагрузке [16].
Диагностический алгоритм
Диагноз ИЛФ основывается на отсутствии известных причин легочного фиброза и наличии картины обычной интерстициальной пневмонии (ОИП) [17]. Даже при наличии гистологической картины ОИП при хирургической биопсии легкого (ХБЛ) окончательный диагноз требует исключения других патологических состояний, ассоциированных с развитием ОИП, включая диффузные заболевания соединительной ткани, пневмокониозы, поражение легких, связанное с приемом лекарственных препаратов, семейный легочный фиброз [18]. При отсутствии данных за альтернативный диагноз, согласно действующим клиническим рекомендациям [4], диагноз ИЛФ устанавливают на основании характерных данных компьютерной томографии высокого разрешения (КТВР) и, при необходимости, результатов биопсии легкого (табл. 3). Следует отметить, что в представленной гистологической классификации выделены "возможный ИЛФ" и "вероятный ИЛФ", когда невозможно однозначно подтвердить или исключить наличие ИЛФ. В таком случае показана повторная оценка данных КТВР и биопсии легкого для уточнения диагноза.
| КТ-картина | Гистологические данные | Диагноз |
|---|---|---|
| ОИП | ОИП | ИЛФ |
| Вероятная ОИП | ||
| Возможная ОИП | ||
| Неклассифицируемый фиброз | ||
| Не соответствует ОИП | Не-ИЛФ | |
| Возможная ОИП | ОИП Вероятная ОИП | ИЛФ |
| Возможная ОИП | Вероятный ИЛФ | |
| Неклассифицируемый фиброз | ||
| Не соответствует ОИП | Не-ИЛФ | |
| Не соответствует ОИП | ОИП | Возможный ИЛФ |
| Вероятная ОИП | Не-ИЛФ | |
| Возможная ОИП | ||
| Неклассифицируемый фиброз | ||
| Не соответствует ОИП |
КТ-диагностика
КТВР играет ключевую роль в диагностике ИЛФ и позволяет установить диагноз приблизительно в 2/3 случаев. В ряде исследований было показано, что КТ-картина типичной ОИП по данным КТВР согласуется с наличием гистологической картины типичной ОИП по данным биопсии легкого в 90-100% случаев [4]. Наличие достоверных КТ-признаков ОИП в настоящее время считают достаточным для диагностики ИЛФ без биопсии легкого. Проведение хирургической биопсии легкого (ХБЛ) рекомендуется при наличии КТ-картины, не типичной для ОИП. В таких случаях диагноз устанавливают на основании сочетания данных КТВР и гистологической картины (табл. 3). Таким образом, точная интерпретация данных КТВР является необходимым условием для постановки диагноза [1].
В настоящее время выделяют три КТ-варианта ОИП "типичная ОИП", которая исключает необходимость проведения ХБЛ, "возможная ОИП" и "не соответствует ОИП". При наличии последних двух вариантов требуется проведение ХБЛ [19].
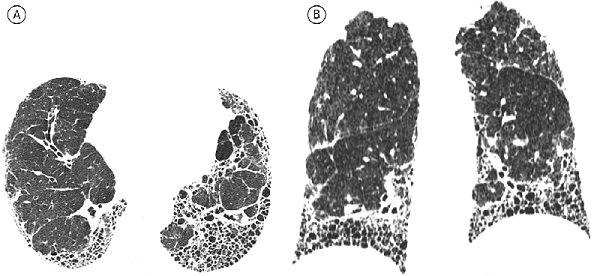
КТ-картина типичной ОИП включает в себя преимущественно базальные и периферические ретикулярные изменения с образованием сотового легкого в сочетании с тракционными бронхоэктазами или без них. Критериями "сотового легкого" считают преимущественно субплевральные кисты диаметром 3-10 мм с четкими, относительно толстыми стенками (1-3 мм), расположенные слоями. Все КТ-признаки, рассматриваемые как "не соответствующие" ОИП, должны отсутствовать (рис. 1). Если все вышеуказанные критерии выполнены, данные КТВР достаточны для диагностики ОИП, а необходимости в проведения биопсии легкого нет [4]. Относительно признаков типичной ОИП заключения разных специалистов обычно хорошо согласуются [20,21]. Тем не менее, следует отметить, что ОИП и ИЛФ не являются синонимами, так как КТизменения, характерные для ОИП, могут отмечаться при ряде других заболеваний, прежде всего диффузных заболеваниях соединительной ткани.
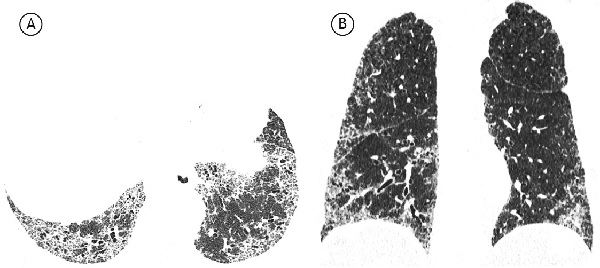
При возможной ОИП наблюдаются преимущественно базальные и периферические ретикулярные изменения без формирования зон сотового легкого. При этом изменения, не соответствующие ОИП, отсутствуют (рис. 2). Картина возможной ОИП менее специфична для ИЛФ, чем картина типичной ОИП. В данном случае дифференциальный диагноз следует проводить, в первую очередь, с неспецифической интерстициальной пневмонией (НСИП), для которой характерны отсутствие участков сотового легкого, преобладание затемнений по типу "матового стекла" над ретикулярными изменениями, относительная сохранность субплевральных зон. Участки сотовой трансформации редко встречаются при НСИП. В одном исследовании они были выявлены менее чем у 5% пациентов с идиопатической НСИП [23].
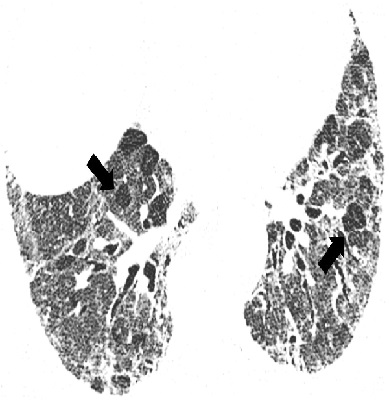
Изменения по данным КТВР, которые считают не соответствующими ОИП, включают в себя следующие: а) преобладание изменений в верхних и средних отделах легких; б) преимущественно перибронховаскулярные изменения; в) значительные по размеру зоны затемнения по типу "матового стекла", распростра ненность которых превышает таковую ретикулярных изменений; г) двусторонние очаговые изменения, преимущественно в верхних отделах легких; д) наличие кист (множественных, двусторонних) вне зон фиброза; е) картина мозаичного затемнения легочной ткани/ наличие "воздушных ловушек" (двусторонние изменения в трех и более долях); ж) наличие зон консолидации (рис. 3).
Несмотря на высокую вероятность наличия ИЛФ при типичной ОИП по данным КТВР, отсутствие зарактерной КТ-картины не должно служить основанием для исключения диагноза ИЛФ [22]. В 2017 г. D. Lynch и соавт. предложили новую КТ-классификацию ОИП, в которой впервые выделена группа неопределенной ОИП (табл. 4) [24].
| Типичная ОИП | Вероятная ОИП | Неопределенная ОИП | Наименее вероятно соответствует ОИП |
|---|---|---|---|
| Преобладание в базальных и субплевральных отделах (редко диффузные изменения); часто неоднородное распределение Зоны "сотового легкого"; ретикулярные изменения с периферическими тракционными бронхо эктазами и бронхиолоэктазами; отсутствие данных за альтернативный диагноз | Преобладание в базальных и субплевральных отделах; часто неоднородное распределение Ретикулярные изменения с периферическими тракционным бронхоэктазами и бронхиолоэктазами; отсутствие зон "сотового легкого"; отсутствие данных за альтернативный диагноз | Вариабельное или диффузное распределение Наличие фиброза в сочетании с небольшими по объему изменениями, не соответствующими ОИП | Преобладание в верхних и средних отделах легких; перибронховаскулярное распределение с относительной сохранностью субплевральных зон Любое из нижеперечисленного: преобладание зон консолидации; значительные по размеру зоны затемнения по типу "матового стекла" (при отсутствии обострения ИЛФ); диффузные очаговые или кистозные изменения; выраженное мозаичное затемнение легочной ткани с наличием "воздушных ловушек" |
Клиническое течение ИЛФ может быть различным. У большинства пациентов отмечается медленно прогрессирующее течение, однако у некоторых пациентов происходит стабилизация патологического процесса, тогда как у других отмечается довольно быстрое прогрессирование заболевания. Что касается выраженности легочных изменений по данным КТВР, то зоны затемнения по типу "матового стекла" чаще всего трансформируются в ретикулярные изменения, которые, в свою очередь, могут прогрессировать и формировать зоны "сотового легкого", размер которых со временем обычно увеличивается. Следует отметить, что общий паттерн легочных изменений также может изменяться: так, КТкартина возможной ОИП может трансформироваться в типичную ОИП [22].
Биопсия легкого
Если однозначные данные о наличии ИЛФ при КТВР отсутствуют, то для подтверждения диагноза показано выполнение хирургической биопсии легких, которую чаще проводят с помощью видеоторакоскопической методики. С целью повышение эффективности биопсия легких должна производиться из разных долей легких. Хотя ХБЛ является наиболее достоверным методом определения гистологической картины ИИП, ее проведение связано с риском возникновения ряда осложнений, наиболее тяжелым из которых является обострение ИЛФ, особенно у пациентов с тяжелой дыхательной и/или сердечной недостаточностью [25]. В связи с этим решение о ее проведении должно приниматься индивидуально с учетом клинической картины, возможных преимуществ для постановки точного диагноза, а также согласия пациента.
В течение последнего десятилетия для гистологического подтверждения диагноза ИЛФ и других вариантов ИИП разработана методика трансбронхиальной криобиопсии легкого (ТБКБЛ). Ее основными преимуществами являются малоинвазивность, отсутствие необходимости в проведении интубации и ингаляционного наркоза и, вследствие этого, низкая частота развития осложнений в сочетании с возможностью получения большого по объему биоптата легкого, достаточного, в абсолютном большинстве случаев, для гистологической верификации диагноза [26]. Так, у пациентов без типичной картины ОИП по данным КТВР проведение ТБКБЛ позволяло установить диагноз приблизительно в 2/3 случаев, что сопоставимо с эффективностью ХБЛ в сходной ситуации [27]. При этом для ТБКБЛ характерны более низкий риск периоперационных осложнений (чаще всего отмечают развитие пневмоторакса и не угрожающего жизни кровотечения в месте проведения биопсии) и смерти, более короткий период госпитализации, что позволяет проводить ТБКБЛ у пациентов с высоким уровнем анестезиологического риска и наличием противопоказаний к ХБЛ [28].Таким образом, внедрение ТБКБЛ в клиническую практику может расширить показания к биопсии легкого и повысить диагностическую точность алгоритма обследования пациентов с подозрением на ИЛФ.
При морфологическом исследовании у пациентов с подозрением на ИЛФ G. Raghu и соавт. выделяют пять возможных гистологических паттернов заболевания (табл. 5) [29,4]. В сочетании с рентгенологическими данными они используются для подтверждения/исключения диагноза ИЛФ (табл. 3) [4,30].
Дифференциальный диагноз
У пациентов с подозрением на ИЛФ должен проводиться тщательный дифференциальный диагноз. При выявлении КТ-картины, соответствующей вероятной или возможной ОИП, что происходит довольно часто, в круг дифференциального диагноза следует включать, в первую очередь, хронический экзогенный аллергический альвеолит и фибротический вариант НСИП. Тем не менее, у части пациентов рекомендованная в данном случае ХБЛ не проводится в связи с наличием противопоказаний (тяжелой дыхательной недостаточности, сопутствующих заболеваний, возрастных ограничений) или нежеланием пациента.
При проведении дифференциального диагноза важно также исключить поражение легких в рамках системного заболевания соединительной ткани, в частности, ревматоидного артрита, системной склеродермии, дерматомиозита, синдрома Шегрена [31], в том числе при наличии КТ-картины типичной ОИП. При наличии у пациента отдельных клинических проявлений или повышения уровня лабораторных аутоиммунных маркеров, не соответствующих конкретному системному заболеванию соединительной ткани, может быть установлен диагноз интерстициальной пневмонии с аутоиммунными чертами [32].
Генетические маркеры ИЛФ
В настоящее время выявлен ряд мутаций и полиморфизмов генов, участвующих в ремоделировании легочной ткани и регуляции врожденного и приобретенного иммунитета, ассоциированных с развитием ИЛФ [33]. К ним относятся, в частности, мутации в генах, кодирующих сурфактантные протеины А и D (S):1–112. P-A и S):1–112. PD), описанные при семейных формах ИЛФ [34]. В ряде исследований выявлена ассоциация генетических полиморфизмов с прогнозом заболевания: в частности, наличие отдельных однонуклеотидных полиморфизмов в гене TLR-3 (Toll-подобный рецептор 3-го типа) ассоциировано с более быстрым прогрессированием заболевания [35]. Также при ИЛФ описан ряд полиморфизмов в генах муцина 5B (MUC5B) и TOLLIP (протеин, взаимодействующий с Toll-подобным рецептором) [36]. Хотя исследование генетических полиморфизмов не является частью диагностического алгоритма при ИЛФ, продолжается поиск генетических маркеров, способных служить предикторами различных вариантов течения заболевания и ответа на терапию.
Обострение ИЛФ
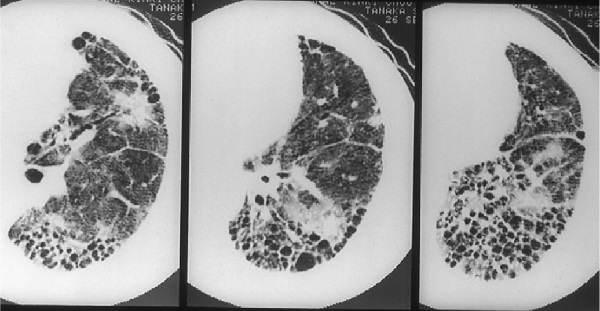
Обострение ИЛФ – это тяжелое жизнеугрожающее состояние, проявляющееся в виде быстрого нарастания дыхательной недостаточности у пациентов с ранее установленным диагнозом ИЛФ [37]. Как правило, характеризуется крайне тяжелым течением; смертность в ряде исследований достигала 85% [38]. В отличие от стабильного или медленно прогрессирующего течения ИЛФ, критерии диагностики его обострения определены менее четко. Согласно данным Н. Collard и соавт. [39], критерии обострения ИЛФ включают в себя наличие предшествующего или впервые выявленного ИЛФ с резким нарастанием одышки, развитием дыхательной недостаточности за предшествующие 30 дней без установленной причины, а также появление новых зон затемнения легочной ткани по типу "матового стекла" и/или консолидации на фоне имевшихся ранее изменений, соответствующих ОИП – зон ретикулярных изменений и "сотового легкого" (рис. 4) [40]. Тем не менее, вышеуказанные критерии обладают низкой специфичностью, в связи с чем при подозрении на обострение ИЛФ должен проводиться дифференциальный диагноз с инфекционным процессом, тромбоэмболией легочной артерии и ее ветвей, пневмотораксом, а также острой левожелудочковой недостаточностью с развитием отека легких [41].
Заключение
Появление новых методов лечения, в частности, антифибротических препаратов, и неэффективность традиционной иммуносупрессивной терапии при ИЛФ подчеркивают важность как можно более ранней постановки диагноза и начала терапии. В течение последнего десятилетия был достигнут значительный прогресс в разработке диагностических алгоритмов для пациентов с ИЛФ. Этому способствовало повышение качества визуализационных методов, более полное понимание роли биопсии легких и разработка гистологических критериев ИЛФ. Все вышеперечисленные параметры должны исследоваться мультидисциплинарной командой специалистов, что в настоящий момент является стандартом диагностики ИЛФ. Несмотря на достигнутые успехи, в диагностике ИЛФ остаются нерешенные вопросы, в основном касающиеся применения инвазивных методов диагностики, в частности, хирургической биопсии легкого. Необходимо продолжать поиск молекулярно-биологических и генетических маркеров ИЛФ и разработку малоинвазивных биопсийных методов для максимально раннего установления диагноза, определения прогноза и разработки стратегии терапии ИЛФ.
Используемые источники
- Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic S):1–112. ociety/European Respiratory S):1–112. ociety statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–48.
- Fernández Pérez ER, Daniels CE, S):1–112. chroeder DR, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis a population-based study. Chest 2010;137:129–37.
- Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006;174:810–6.
- Raghu G, Collard HR, Egan JJ, et al. An Official ATS):1–112. /ERS):1–112. /JRS):1–112. /ALAT S):1–112. tatement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824.
- Wolters PJ, Blackwell TS):1–112. , Eickelberg O, et al. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir Med 2018; 6:154–60.
- King TE, Tooze J, S):1–112. chwarz MI, et a. Predicting survival in idiopathic pulmonary fibrosis: scoring system and survival model. Am J Respir Crit Care Med 2001; 164:1171–81.
- Фомин В.В., Попова Е.Н., Лебедева М.В. Идиопатический легочный фиб- роз: близки ли мы к общепринятым стандартам диагностики и лечения? Фарматека 2012;5:10–4.
- Armanios M, Alder JK, Chen JJ-L, et al. S):1–112. hort telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad S):1–112. ci US):1–112. A 2008;105:13051–6.
- Leslie KO. Idiopathic pulmonary fibrosis may be a disease of recurrent, tractional injury to the periphery of the aging lung: A unifying hypothesis regarding etiology and pathogenesis. Arch Pathol Lab Med 2012;136:591–600.
- Blackwell TS):1–112. , Tager AM, Borok Z, et al. Future directions in idiopathic pul- monary fibrosis research an NHLBI workshop report. Am J Respir Crit Care Med 2014;189:214–22.
- Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetyl- cysteine for pulmonary fibrosis. N Engl J Med 2012;366:1968–77.
- Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS):1–112. /ERS):1–112. /JRS):1–112. /ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 2015; 192:e3–19.
- Yusen RD, S):1–112. hearon TH, Qian Y, et al. Lung transplantation in the United S):1–112. tates, 1999-2008: S):1–112. pecial feature. Am J Transplant 2010;10:1047–68.
- Авдеев С.Н. Идиопатический легочный фиброз: современная концепция и подходы к диагностике. Практическая пульмонология 2014;4:16–23.
- American Thoracic S):1–112. ociety. Idiopathic pulmonary fibrosis: diagnosis and treat- ment. International consensus statement. American Thoracic S):1–112. ociety (ATS):1–112. ), and the European Respiratory S):1–112. ociety (ERS):1–112. ). Am J Respir Crit Care Med 2000; 161:646–64.
- Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac S):1–112. oc 2006;3:315–21.
- Cepika A-M, Marinic I, Morovic-Vergles J, et al. Effect of steroids on the fre- quency of regulatory T сells and expression of FOXP3 in a patient with systemic lupus erythematosus: a two-year follow-up. Lupus 2007;16:374–7.
- Baldi BG, Pereira CA, Rubin AS):1–112. , et al. Highlights of the Brazilian Thoracic Association guidelines for interstitial lung diseases. J Bras Pneumol 2012;38(3): 282-91.
- Визель А.А., Визель И.Ю. Идиопатический легочный фиброз: состояние проблемы. Вестник современной клинической медицины 2017;10:14–21.
- Hodnett PA, Naidich DP. Fibrosing interstitial lung disease: A practical high-res- olution computed tomography-based approach to diagnosis and management and a review of the literature. Am J Respir Crit Care Med 2013;188:141–9.
- Flaherty KR, Thwaite EL, Kazerooni EA, et al. Radiological versus histological diagnosis in UIP and NS):1–112. IP: S):1–112. urvival implications. Thorax 2003;58:143–8.
- Paulo P, Torres S):1–112. , Rabahi MF, et al. Usual interstitial pneumonia: typical, possi- ble, and ‘inconsistent’ patterns. J Bras Pneumol 2017;43:393–8.
- Travis WD, Hunninghake G, King TE, et al. Idiopathic nonspecific interstitial pneumonia: Report of an American Thoracic S):1–112. ociety Project. Am J Respir Crit Care Med 2008;177:1338–47.
- Lynch DA, S):1–112. verzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pul- monary fibrosis: A Fleischner S):1–112. ociety White Paper. Lancet Respir Med 2017;6: 138–53.
- Kolb M, S):1–112. hargall Y. Lung surgery in interstitial lung disease - a safe and useful procedure? J Thorac Dis 2013;5:375–7.
- Bango-Аlvarez A, Ariza-Prota M, Torres-Rivas H, et al. Transbronchial cryobiop- sy in interstitial lung disease: experience in 106 cases – how to do it. ERJ Open Res 2017;3:00148–2016.
- Lentz RJ, Christine Argento A, Colby TV, et al. Transbronchial cryobiopsy for diffuse parenchymal lung disease: A state-of-the-art review of procedural tech- niques, current evidence, and future challenges. J Thorac Dis 2017;9:2186–203.
- Tomassetti S):1–112. , Wells AU, Costabel U, et al. Bronchoscopic lung cryobiopsy increases diagnostic confidence in the multidisciplinary diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016;193:745–52.
- Baddini-Martinez J, Baldi BG, Costa CH, et al. Update on diagnosis and treat- ment of idiopathic pulmonary fibrosis. J Bras Pneumol 2015;41:454–66.
- Цветкова О.А., Воронкова О.О. Современный подход к терапии больных идиопатическим легочным фиброзом. Клиническая медицина 2017;95: 281–5.
- Авдеев С.Н. Идиопатический легочный фиброз: современные подходы к терапии. Практическая пульмонология 2015;1 22–31.
- Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory S):1–112. ociety/American Thoracic S):1–112. ociety research statement: Interstitial pneumonia with autoimmune features. Eur Respir J 2015;46:976–87.
- Brownell R, Kaminski N, Woodruff PG, et al. Precision medicine: The new fron- tier in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016;193: 1213–8.
- Thomas AQ, Lane K, Phillips J, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspe- cific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002; 165:1322–8.
- O’Dwyer DN, Armstrong ME, Trujillo G, et al. The toll-like receptor 3 L412F polymorphism and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2013;188:1442–50.
- Oldham JM, Ma S):1–112. F, Martinez FJ, et al. TOLLIP, MUC5B, and the response to N-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2015;192:1475–82.
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pul- monary fibrosis an international working group report. Am J Respir Crit Care Med 2016;194:265–75.
- Hambly N, Cox G, Kolb M. Acute exacerbations of idiopathic pulmonary fibro- sis: tough to define; tougher to manage. Eur Respir J 2017;49:1700811.
- Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2007;176:636–43. '
- Akira M, Kozuka T, Yamamoto S):1–112. , S):1–112. akatani M. Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008;178:372–8.
- Juarez MM, Chan AL, Norris AG, et al. Acute exacerbation of idiopathic pul- monary fibrosis-A review of current and novel pharmacotherapies. J Thorac Dis 2015;7:499–519