Клиническое значение определения экскреции с мочой нефрина и подоцина у больных сахарным диабетом
Изучение значения экскреции с мочой маркеров подоцитарного повреждения для ранней диагностики диабетической нефропатии (ДН) у больных сахарным диабетом (СД) с разной выраженностью альбуминурии/протеинурии.
Обследовано 74 больных СД 1 и 2 типа (у 33 и 44, соответственно), которые были распределены на 3 группы: 1-я (n=41) – альбуминурия <30 мг/г креатинина мочи (A1), 2-я (n=13) – альбуминурия 30-300 мг/г (А2), 3-я (n=20) – протеинурия. Хроническая болезнь почек (ХБП) 1 стадии выявлена у 41 пациента, ХБП 2 стадии – у 25, ХБП 3 стадии – у 8. Артериальная гипертония (АГ) наблюдалась у 52 (70%) из 74 обследованных и чаще встречалась у больных СД 2 типа. Контрольную группу составили 10 здоровых добровольцев. Содержание структурных белков подоцитов (нефрина и подоцина) в моче определяли иммуноферментным методом.
Нефринурия >5,84 нг/мл/г отсутствовала у здоровых добровольцев и определялась у 63%, 77% и 80% пациентов трех групп, соответственно, подоцинурия >1,73 нг/мл/г – у 78%, 54% и 83%. У пациентов с протеинурией экскреция нефрина с мочой была достоверно выше, чем у больных с альбуминурией <30 мг/г, в то время как подоцинурия была одинаково высокой в трех группах и не отличалась у пациентов с СД 1 и 2 типа. Выявлена прямая корреляция нефринурии с альбуминурией (r=0,947 р<0,05). Нефринурия и подоцинурия прямо коррелировали с сывороточным уровнем креатинина (r=0,489 р<0,05 и r=0,468 р<0,05) и обратно – cо скоростью клубочковой фильтрации (r=-0,461 р<0,05 и r=-0,36 р<0,05). При длительности СД менее 5 лет нефринурия прямо коррелировала с уровнем НbА1c , а у больных СД 2 типа – обратно с систолическим АД.
Определение уровней в моче нефрина и подоцина может использоваться для ранней диагностики и мониторирования течения ДН.
Сахарный диабет (СД) – хроническое неинфекционное заболевание, темпы роста распространенности которого приобрели масштаб мировой эпидемии. В мире 415 миллионов людей в возрасте 20–79 лет страдают СД, а к 2040 г. прогнозируют увеличение их количества еще на 55% – до 640 миллионов [1]. По данным Российского государственного регистра больных СД, в конце 2016 г. в нашей стране насчитывалось 4 348 422 пациентов (3% населения РФ) [2]. Однако эти данные недооценивают реальное количество больных СД, поскольку учитывают только выявленные и зарегистрированные случаи заболевания. Результаты масштабного Национального эпидемиологического исследования (NATION) показали, что в обычной клинической практике диагностируется менее 50% пациентов с СД 2 типа [3]. Установленная при активном скрининге в разных возрастных группах истинная распространенность СД в России приблизительно в 2-3 раза превысила официально зарегистрированную и составила около 10 миллионов человек (7% населения нашей страны).
Колоссальные экономические расходы, социальный ущерб, высокая инвалидизация и смертность при СД связаны с развитием сосудистых осложнений, в частности, диабетической нефропатии (ДН), занимающей ведущие позиции в структуре причин терминальной почечной недостаточности [1,4,5]. В США и странах Европы на долю СД приходится до 44-46,5% больных, получающих заместительную почечную терапию.
В силу прогрессирующего характера течения ДН и ограниченных возможностей ее лечения на поздних стадиях особую актуальность приобретает раннее выявление нефропатии на этапе потенциально обратимых изменений в почках с целью своевременного начала нефропротективной терапии. Единственным используемым в обычной клинической практике методом ранней диагностики ДН является определение альбуминурии, которая коррелирует с выраженностью морфологических изменений в почках [6]. Экскреция альбумина с мочой от 30 до 300 мг/сут (30-300 мг/г креатинина мочи), которую ранее называли микроальбуминурией, длительное время считалась маркером начальных функциональных и структурных изменений в почке при СД. Однако проведенные к настоящему времени исследования доказали, что характерные для СД экспансия мезангия, увеличение толщины гломерулярной базальной мембраны и объема клубочков обнаруживаются и при меньших значениях альбуминурии (<30 мг/сут или <30 мг/г креатинина мочи). При наличии микроальбуминурии эти изменения более выражены, в части клубочков уже может определяться гломерулосклероз, а появление стойкой протеинурии свидетельствует о потере около 50–70% клубочков [5-8]. Сегодня микроальбуминурию называют “высокой альбуминурией” (подчеркивая, что это уже клинически значимый, а не самый ранний признак), для альбуминурии <30 мг/сут, традиционно считавшейся “нормоальбуминурией”, предложен термин “повышенная альбуминурия”, а “оптимальной” считают альбуминурию <10 мг/сут [8-10]. Убедительно доказано, что альбуминурия 30-300 мг/сут и даже 10-30 мг/сут достоверно ассоциируется с повышением риска развития сердечно-сосудистых катастроф и общей смертности даже при отсутствии СД и артериальной гипертонии, в то время как у больных СД эта связь еще более тесная. Поэтому значение повышения альбуминурии для диагностики субклинической ДН далеко неабсолютное.
Более информативными и специфичными ранними маркерами поражения почек при СД могут быть структурно-функциональные белки подоцитов – ключевых компонентов гломерулярного фильтра [11,12]. Целью исследования было изучение значения экскреции с мочой маркеров подоцитарного повреждения (нефрина и подоцина) для ранней диагностики ДН у больных СД с разной выраженностью альбуминурии/протеинурии.
Материал и методы
В исследование были включены 74 пациента с СД, в том числе 30 – с СД 1 типа и 44 – с СД 2 типа. Среди них было 37 мужчин и 37 женщин в возрасте от 18 до 80 лет. Средний возраст пациентов с СД 1 и 2 типа типа составил 36,9±17 лет и 60,5±10 лет, соответственно, длительность заболевания – 18,4±13 лет и 11,5±7,5 лет. Контрольную группу составили 10 здоровых добровольцев, в том числе 4 мужчины и 6 женщин в возрасте от 19 до 77 лет.
Из исследования исключали пациентов с декомпенсированным СД (уровень гликозилированного гемоглобина HbA1c >10%), протеинурией более 2 г/сут, СКФ<15 мл/мин/1,73 м2, острым воспалительным заболеванием любой этиологии на момент исследования, острым или хроническим гломерулонефритом, ишемической болезнью почек, злокачественной опухолью, системными васкулитами, амилоидозом, сердечной недостаточностью III-IV функционального класса (по NYHA), острым нарушением мозгового кровообращения, тяжелой артериальной гипертонией, а также беременных женщин.
У всех больных определяли концентрацию альбумина в моче иммунотурбидиметрическим методом и рассчитывали соотношение концентрации альбумина к креатинину мочи. СКФ рассчитывали по формуле CKD-EPI. Содержание нефрина (n=74) и подоцина (n=70) в утренней порции мочи измеряли с помощью наборов для иммуноферментного анализа (ELISA Kit) производства CUSABIO (China) с расчетом концентрации на единицу креатинина мочи в исследованном образце.
Статистическая обработка полученных данных проведена с использованием компьютерной программы STATISTICA 10.0. При анализе уровней медиаторов в моче, имеющих отличное от нормального распределение, оценивали медиану и интерквартильный размах [25%; 75%]. При сравнении двух групп использовали непараметрический критерий Манна-Уитни. Для оценки связей между исследуемыми показателями применяли непараметрический корреляционный анализ по методу Спирмена. Достоверность различий частот в двух группах оценивали с помощью критерия χ2 с поправкой Йетса.
Результаты
Содержание маркеров подоцитарного повреждения сравнивали в трех группах больных СД, различавшихся по выраженности альбуминурии/протеинурии [8]: 1-я (n=41) – альбуминурия <30 мг/г креатинина мочи (A1), 2-я (n=13) – альбуминурия 30-300 мг/г (А2), 3-я (n=20) – протеинурия. Артериальная гипертония наблюдалась у 52 (70%) из 74 обследованных, преимущественно у больных СД 2 типа. Для лечения артериальной гипертонии применяли ингибиторы АПФ и блокаторы кальциевых каналов. ХБП 1 стадии выявлена у 41 пациента, ХБП 2 стадии – у 25, ХБП 3 стадии – у 8.
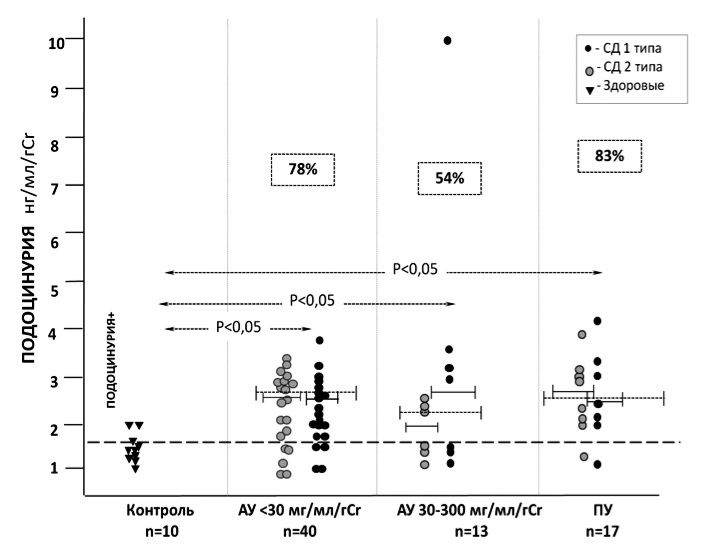
Учитывали нефринурию и подоцинурию, превышавшие 75 перцентиль в контрольной группе (т.е. практически не встречающиеся у здоровых людей): >5,84 нг/мл/г и >1,73 нг/мл/г, соответственно. Нефринурия определялась у 69% (52/74) больных СД, в том числе у 63% (26/41) пациентов 1-й группы, у 77% (10/13) – 2-й группы и у 80% (16/20) – 3-й группы (рис. 1). Подоцинурия была выявлена у 74% (31/70) больных СД, в том числе у 78% (31/40) пациентов 1-й группы, у 54% (7/13) – 2-й группы и у 83% (52/70) – 3-й группы (рис. 2). Соотношение больных СД 1 и СД 2 типа с нефринурией и подоцинурией в каждой группе было практически одинаковым.
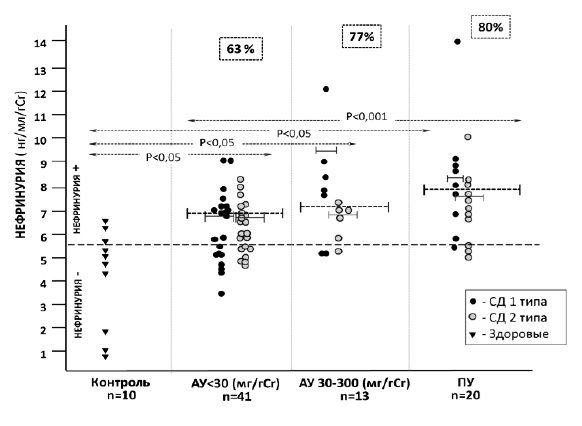
Средний уровень нефрина в моче у больных СД 1 и 2 типа достоверно не отличался в 1-й группе (7,06 [5,98;7,22] и 7,03 [6,07;7,82] нг/мл/г, соответственно; р>0,05) и 2-й группе (9,56 [7,66;9,56] и 6,91 [6,73;7,06] нг/мл/г; р>0,05), что, по-видимому, отражает общность механизмов повреждения подоцитов при диабете. При клинически явной ДН, протекающей с протеинурией, экскреция нефрина с мочой была достоверно выше, чем у пациентов с альбуминурией А1 (табл. 1). У пациентов с СД 1 типа и протеинурией экскреция нефрина с мочой достоверно превышала таковую у больных СД 2 типа (8,67 [8,04;9,93] и 7,46 [7,38;7,68] нг/мл/г, соответственно, р<0,05).
| Нефринурия (нг/мл/г) | Подоцинурия (нг/мл/г) | |
|---|---|---|
| Примечание: приведены медианы и интерквартильный размах | ||
| Больные СД | ||
| Альбуминурия <30 мг/г (I) | 7,04 [6,07;7,22] | 2,50 [2,03;3,01] |
| n=26 | n=31 | |
| Альбуминурия 30-300 мг/г (II) | 7,36 [6,73;9,56] | 2,03 [1,48;3,04 |
| n=10 | n=7 | |
| Протеинурия (III) | 7,76 [7,42;8,49] | 2,70 [2,03;3,73] |
| n=16 | n=14 | |
| Здоровые (IV) | 5,3 [1,0;5,84] | 1,56 [1,21;1,73 |
| P | pI-III <0,001 | pI-III >0,05 |
| pI-IV <0,05 | pI-IV <0,05 | |
| pI-II >0,05 | pI-II >0,05 | |
| pII-IV <0,05 | pII-IV <0,05 | |
| pII-III >0,05 | pII-III >0,05 | |
| pIII-IV <0,05 | pIII-IV <0,05 | |
Средний уровень подоцина в моче в трех группах был одинаково высоким (табл. 1) и не отличался у пациентов с СД 1 и 2 типа: 2,50 [2,10;2,86] и 2,70 [2,40;3,01] нг/мл у пациентов с СД 1 и 2 типа и альбуминурией А1, соответственно (р>0,05); 2,05 [2,03;2,45] и 2,60 [2,0;3,27] нг/мл/г у пациентов с СД 1 и 2 типа и альбуминурией А2 (р>0,05); 2,60 [2,12;3,09] и 2,80 [2,20;4,24] нг/мл/г у пациентов с СД 1 и 2 типа и протеинурией (р>0,05).
У пациентов с СД 1 типа выявлена прямая корреляция нефринурии и альбуминурии (r=0,47; р<0,05), которая была более выраженной у больных с альбуминурией А2 (r=0,947; р<0,05). Наиболее сильной и достоверной была взаимосвязь нефринурии с протеинурией (r=0,953; р<0,01).
Нефринурия и подоцинурия у больных СД 1 типа прямо коррелировали с сывороточным уровнем креатинина сыворотки крови (r=0,489; р<0,05 и r=0,468; р<0,05, соответственно) и обратно – с расчетной СКФ (r=-0,461; р<0,05 и r=-0,36; р<0,05, соответственно), что отражает роль подоцитарного повреждения не только в нарушении проницаемости гломерулярного фильтра, но и в развитии гломерулосклероза и формировании дисфункции почек. На это указывает и выявленная нами прямая взаимосвязь между нефринурией и длительностью СД, которая была достоверной и имела наибольшую силу у пациентов с расчетной СКФ менее 60 мл/мин/1,73 м2, независимо от типа диабета (r=0,73; р<0,05). У пациентов с длительностью СД 1 и СД 2 менее 5 лет нефринурия прямо коррелировала с концентрацией НbА1c (r=0,84; р<0,01).
На величину нефринурии при разной длительности СД оказывала влияние АГ, особенно у больных СД 2 типа, у которых она часто предшествовала развитию патологии почек. У данной категории больных нами выявлена прямая достоверная связь систолического АД с экскрецией нефрина с мочой (r=0,33; р<0,05).
Обсуждение
Современные достижения молекулярной медицины и экспериментальной нефрологии позволили расширить представления о механизмах, приводящих к развитию альбуминурии и протеинурии. Подтверждена ключевая роль в этих процессах подоцитов – основных компонентов щелевой диафрагмы клубочков [13-17]. Сложное структурное устройство подоцита обеспечивает широкий набор его функций и приспособительных реакций в физиологических условиях, но в то же время делает эту клетку очень чувствительной к повреждению.
После воздействия различных патогенных факторов, ассоциированных с СД (гипергликемии, конечных продуктов гликозилирования, компонентов активированной ренин-ангиотензин-альдостероновой системы [РААС], внутриклубочковой гипертонии, оксидатив ного стресса и др.), подоциты подвергаются структур но-функциональным изменениям (так называемая “подоцитопатия”). Признаками подоцитопатии являются сглаживание ножек подоцитов с нарушением проницаемости щелевидной диафрагмы, гипертрофия, апоптоз, отслоение подоцитов от базальной мембраны клубочка со слущиванием их в мочевое пространство и появлением в моче как целых клеток (подоцитурия), так и структурных белков (нефринурия), уменьшение количества подоцитов в клубочке (подоцитопения) [12,18-23]. Ультраструктурные и функциональные нарушения в подоцитах предшествуют повышению альбуминурии и могут обнаруживаться даже при непродолжительном течении СД, что определяет еще один важный аспект изучения маркеров подоцитарной дисфункции – для ранней диагностики и мониторирования течения ДН.
По нашим данным, высокая экскреция маркеров подоцитарного повреждения отмечалась у части пациентов с альбуминурией <30 мг/г, а при развитии протеинурии высокие нефринурия и подоцинурия встречались чаще. Самая высокая нефринурия определялась у пациентов с протеинурией, что отражало болеевыраженные изменения в подоцитах при клинически явной ДН. Полу ченные нами результаты согласуются с данными других исследований, свидетельствующими о вовлечении подоцитов в процессы инициации почечного повреждения при СД. В частности, А. Patari и соавт. [24] в исследовании, проводившимся методом поперечного среза, выявили нефринурию методом иммуноблоттинга у 30% больных СД 1 типа с нормоальбуминурией (в нашем исследовании соответствует альбуминурии А1), у 17% – с микроальбуминурией (в нашем исследовании соответствует альбуминурии А2) и у 28% – с протеинурией, тогда как в моче здоровых добровольцев нефрин не определялся. В. Jim и соавт. [25] обнаружили нефринурию у 54% больных с нормоальбуминурией и у всех больных СД 2 типа с протеинурией и микроальбуминурией. Как и в нашем исследовании, средняя экс кре ция нефрина с мочой у пациентов с микроальбуминурией и особенно протеинурией достоверно превышала таковую у пациентов с меньшей альбуминурией.
Повышенные концентрации глюкозы, воздействуя на подоциты, активируют экспрессию в них коллагена IV типа и фибронектина, подавляют механизмы расщепления внеклеточного матрикса (снижают продукцию матриксной металлопротеиназы 2 типа и увеличивают продукцию ее тканевого ингибитора), что приводит к утолщению базальной мембраны клубочков [20]. Гипергликемия вызывает реорганизацию актиновых фибрилл в цитоскелете подоцита с нарушением архитектоники его тела и ножек, способствуя таким образом развитию альбуминурии/протеинурии [20]. Одним из основных механизмов, посредством которых гипергликемия оказывает повреждающее действие на структуры почки, в том числе подоциты, является неферментативное гликозилирование белков с образованием токсичных продуктов. Наибольшее повреждающее действие оказывают конечные продукты гликозилирования. Они образуются в организме больного СД в течение нескольких месяцев, после чего даже тщательная компенсация гликемии уже не способна полностью устранить присутствие этих веществ. Именно необратимостью конечных продуктов гликозилирования объясняют продолжающееся прогрессирование сосудистых осложнений даже при хорошей компенсации СД. Подоциты являются мишенью для конечных продуктов гликозилирования, о чем свидетельствует экспрессия ими соответствующих рецепторов. Так, in vitro было продемонстрировано снижение экспрессии нефрина подоцитами экспериментальных животных с СД и пациентов с СД под действием гликированного альбумина, эффект которого проявлялся при взаимодействии с рецепторами конечных продуктов гликозилирования [26]. Экспрессию рецепторов конечных продуктов гликозилирования в подоцитах активирует не только гипергликемия, но и ангиотензин II через АТ2-рецепторы [26]. Эти сигнальные пути могут представлять дополнительный интерес как потенциальный объект воздействия препаратов, блокирующих РААС, с точки зрения уменьшения токсических эффектов конечных продуктов гликозилирования.
В нашем исследовании у пациентов с СД 1 и СД 2 типа с длительностью заболевания не более 5 лет определялась сильная и высоко достоверная взаимосвязь нефринурии с уровнем гликозилированного гемоглобина, что также указывает на ключевую роль гипергликемии в формировании подоцитарной дисфункции даже при непродолжительном течении СД и подчеркивает необходимость достижения оптимального контроля гликемии для профилактики развития и нарастания тяжести ДН. По современным представлениям, повреждающее воздействие на подоциты оказывает системная, а также внутриклубочковая гипертония, что было продемонстрировано при ДН и гипертоническом нефроангиосклерозе [27-30]. Установлено, что длительное воздействие мощного гидравлического пресса приводит к механическому растяжению подоцитов, нарушению синтеза адгезивных белков, снижению экспрессии ряда структурных подоцитарных белков [31]. Введение ангиотензина II крысам помимо развития артериальной гипертонии сопровождалось апоптозом подоцитов и уменьшением экспрессии нефрина [32]. Кроме того, в эксперименте показано, что подоциты сами являются одним из источников синтеза компонентов РААС в почке. Высокие концентрации глюкозы и механическое растяжение индуцируют синтез ангиотензина II подоцитами через активацию экспрессии ангиотензиногена. Под действием повреждающих факторов подоциты экспрессируют АТ1- и АТ2-рецепторы, приобретая, таким образом, способность отвечать на действие циркулирующего ангиотензина II. Помимо гипергликемии и механического растяжения продукцию ангиотензина II в подоцитах активируют трансформирующий фактор роста β1, реактивные кислородные радикалы, компоненты белков, выделяющихся с мочой [33,34].
В нашем исследовании достоверная взаимосвязь артериальной гипертонии с выраженностью экскреции нефрина с мочой определялась у больных СД 2 типа, в то время как при СД 1 типа она не достигла статистической значимости, что, как мы полагаем, связано с большей частотой артериальной гипертонии именно у больных СД 2 типа. Больные СД 2 типа были старше, многие из них уже имели АГ к моменту развития ДН.
Экспериментальными исследованиям последних лет убедительно доказано, что повреждение подоцитов играет важную роль не только в нарушении проницаемости фильтрационного барьера и развитии протеинурии, но и в формировании гломерулосклероза и нарушении функции почек [15]. При интенсивном или продолжительном воздействии повреждающих факторов происходит значительное слущивание подоцитов в мочевое пространство, что при ограниченной пролиферативной способности этих клеток приводит к подоцитопении. На месте потери подоцита базальная мембрана клубочков оголяется и сращивается с капсулой Шумлянского-Боумена, формируя очаги гломерулосклероза. Кроме того, в процессе повреждения подоциты утрачивают способность экспрессировать специфические подоцитарные белки, меняют эпителиальный фенотип и начинают экспрессировать маркеры мезенхимальных клеток. Подобно фибробластам, трансдифференцированные подоциты приобретают способность продуцировать матриксные белки (фибронектин, коллаген и др.), ускоряя, таким образом, формирование гломерулосклероза и нарушение функции почек [15,34,35].
На важную роль подоцитарной дисфункции в механизмах прогрессирования ДН косвенно указывают и результаты нашего исследования. Так, у пациентов с СД с разной выраженностью альбуминурии/протеинурии экскреция с мочой нефрина прямо коррелировала с уровнем креатинина сыворотки крови и обратно – с СКФ, а у пациентов со стойким снижением СКФ менее 60 мл/мин/1,73 м2 отмечалась сильная прямая взаимосвязь между нефринурией и длительностью СД, что отражает причинно-следственную взаимосвязь пролонгированного повреждения подоцитов при длительном течении СД с развитием дисфункции почек.
Заключение
Современные скрининговые тесты позволяют выявить ДН только на стадии клинически значимой альбуминурии (А1-А2), хотя начальные структурные и функциональные нарушения в почках развиваются задолго до повышения экскреции с мочой альбумина. В нашем исследовании было показано, что у большинства (от 63 до 74%) больных СД определяется достоверное увеличение уровня в моче маркеров повреждения подоцитов (нефрина, подоцина), предшествующее развитию клинически значимой альбуминурии и протеинурии. С учетом результатов предыдущих экспериментальных исследований полученные данные указывает на возможность использования данных мочевых тестов для ранней диагностики гломерулярного повреждения при СД. Тесные корреляции уровней биомаркеров подоцитарной дисфункции (в большей степени нефринурии) в моче с клиническими проявлениями поражения почек (выраженностью альбуминурии/протеинурии, артериальной гипертонией, почечной дисфункцией), а также с уровнем гликозилированного гемоглобина крови указывают на перспективы применения изученных мочевых тестов для неинвазивного мониторирования развивающихся при СД гломерулярных изменений и оценки риска их прогрессирования. Необходимы дальнейшие более крупные исследования с целью определения диагностических концентраций нефрина и подоцина и оценки чувствительности и специфичности данных мочевых тестов в качестве ранних маркеров поражения почек при СД.
Используемые источники
- International Diabetes Federation atlas (7th edition update). Brussels, Belgium. International Diabetes Federation; 2015. Available from: http://www.diabetesatlas.org/
- Дедов И.И., Шестакова М.В., Викулова О.К. Эпидемиология сахарного диабета в Российской Федерации: клинико-статистический анализ по данным Федерального регистра сахарного диабета. Сахарный диабет 2017; 20(1):13-41.
- Дедов И.И., Шестакова М.В., Галстян Г.Р. Распространенность сахарного диабета 2 типа у взрослого населения России (исследование NATION). Сахарный диабет 2016;19(2):104-12.
- Дедов И.И. Сахарный диабет: развитие технологий в диагностике, лечении и профилактике. Сахарный диабет 2010;13(3):6-13.
- Шестакова М.В., Шамхалова М.Ш., Ярек-Мартынова И.Я. и др. Сахарный диабет и хроническая болезнь почек: достижения, нерешенные проблемы и перспективы лечения. Сахарный диабет 2011;14(1):81-8.
- Shestakova M, Mukhin N, Dedov I, et al. Protein-loading test, urinary albumin excretion and renal morphology in diagnosis of subclinical diabetic nephropathy. J Int Med 1992;231(3):213-7.
- Dalla Vestra M, Saller A, Bortoloso E, et al. Structural involvement in type 1 and type 2 diabetic nephropathy. Diabetes Metab 2000;26(4):8-14.
- Смирнов А.В., Добронравов В.А, Кисина А.А. и др. Клинические рекомендации по диагностике и лечению диабетической нефропатии. Нефрология 2015;19(1):67-77.
- National Kidney Foundation. KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 update. Am J Kidney Dis 2012;60(5):850-86.
- KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int 2013;3:1-150.
- Lee SY, Choi ME. Urinary biomarkers for early diabetic nephropathy: beyond albuminuria. Pediatr Nephrol 2015;30(7):1063–75.
- Бобкова И.Н., Шестакова М.В., Щукина А.А. Диабетическая нефропатия – фокус на повреждение подоцитов. Нефрология 2015;2(19):33-44.
- Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev 2003;83(1):253-307.
- Barisoni L, Kopp JB. Update in podocyte biology: putting one's best foot forward. Curr Opin Nephrol Hypertens 2003;12(3):251-8.
- Shankland SJ. The podocyte′s response to injury: Role in proteinuria and glomerulosclerosis. Kidney Int 2006:69;2131-47.
- Kriz W, Elger M, Nagata M, et al. The role of podocytes in the development of glomerular sclerosis. Kidney Int 1994;suppl 45;S64-S72
- Granier C, Makni K, Molina L. Gene and protein markers of diabetic nephropathy. Nephrol Dial Transplant 2008;23:7922-799.
- Ziyadeh FN, Wolf G. Pathogenesis of podocytopathy and proteiuria in diabetic glomerulopathy. Curr Diab Rev 2008;4:39-45.
- Wolf G, Chen S, Ziyadeh FN. From the periphery of glomerular capillary wall toward the center of disease. Podocyte injury comes of age in diabetic nephropathy. Diabetes 2005;54(6):1626-34.
- Stitt-Cavanagh E, MacLeod L, Kennedy CRJ. The podocyte in diabetic kidney disease. Scient World J 2009; 9:1127-39.
- Reddy GR, Kotlyarevska K, Ransom RF. The podocyte and diabetes mellitus: is the podocyte key to the origins of diabetic nephropathy? Сurr Opin Nephrol Hypertens 2008;17:32-6.
- Steffes MW, Schmidt D, McGregory R, Basgen JM. Glomerular cell number in normal subject and in type 1diabetic patients. Kidney Int 2001;59:2104-13.
- Pätäri A, Forsblom C, Havana R, et al. Nephrinuria in diabetic nephropathy of type 1 diabetes. Diabetes 2003;52:2969-74.
- Jim B, Ghanta M, Qipo A, et al. Dysregulated nephrin in diabetic nephropathy of type 2 diabetes: A cross Sectional study. PLoS ONE 2012;7(5):e36041.
- Gruden G, Perin PC, Camussi G. Insight on the pathogenesis of diabetic nephropathy from the study of podocyte and mesangial cell biology. Curr Diab Rev 2005;1(1):27-40.
- Cooper ME. Interaction of metabolic and haemodinamic factors in mediating experimental diabetic nephropathy. Diabetologia 2001;44:1957-72.
- Wang G, Lai FM, Kwan BC, et al. Podocyte loss in human hypertensive nephrosclerosis. Am J Hypertens 2009;22:300-6.
- Forbes JM, Bonnet F, Russo LM, et al. Modulation of nephrin in the diabetic kidney: association with systemic hypertension and increasing albuminuria. J Hypert 2002;20(5):985-92.
- Amazonas RB, de Almedia Sanita R, Kawashi H, et al. Prevention of hypertension with or without renin-angiotensin system inhibition precludes nephrin loss in the early stage of experimental diabetes mellitus. Nephron Physiol 2007;107:57-64.
- Lewko B, Stepinski J. Hypergliycemia and mechanical stress: Targeting the renal podocyte. J Cell Physiol 2009;221(2):288-95.
- Jia J, Ding G, Zhu J, et al. Angiotensin II infusion induced nephrin expression changes and podocyte apoptosis. Am J Nephrol 2008;28(3):500-7.
- Abbate M, Zoja C, Morigi M, et al. Transforming growth factor-beta1 is up-regulated by podocytes in response to excess intraglomerular passage of proteins: a central pathway in progressive glomerulosclerosis. Am J Pathol 2002;161(6): 2179–93.
- Durvasula RV, Petermann AT, Hiromura K, et al. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int 2004;65(1):30–9.
- Li Y, Kang YS, Dai C, et al. Epithelial-to-mesenchymal transition is a potential pathway leading to podocyte dysfunction and proteinuria. Am J Pathol 2008; 172(2):299–308.
- Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol 2010;21:212-2