Интерстициальная пневмония с аутоиммунными признаками (ИПАП): мультидисциплинарный диагноз в пульмонологии и ревматологии
Диагноз идиопатической интерстициальной пневмонии с аутоиммунными признаками (ИПАП) объединяет пациентов с идиопатической интерстициальной пневмонией и рядом клинических, серологических и/или морфологических проявлений, указывающих на наличие системного аутоиммунного процесса, который, однако, не соответствует современным критериям определенного системного заболевания соединительной ткани. Результаты клинических исследований указывают на значительную неоднородность больных данной группы. В обзоре литературы обсуждаются классификационные критерии ИПАП, а также дальнейшие перспективы их применения для улучшения диагностики и лечения пациентов с данной патологией.
Идиопатические интерстициальные пневмонии (ИИП) представляют собой группу диффузных воспалительных и/или фибротических заболеваний легких, объединенных на основании сходных клинических, рентгенологических и гистологических признаков. Диагноз ИИП требует исключения известных причин интерстициальной пневмонии, таких как воздействие экзогенных факторов, токсические эффекты лекарственных препаратов или системное заболевание соединительной ткани (СЗСТ) [1]. Последние представляют собой группу аутоиммунных заболеваний, таких как ревматоидный артрит, системная красная волчанка, идиопатические воспалительные миопатии, синдром Шегрена, системная склеродермия и смешанное заболевание соединительной ткани. Интерстициальная пневмония (ИП) может быть одним из проявлений СЗСТ, причем в части случаев ее развитие предшествует появлению других симптомов заболевания [2-5].
У пациентов с ИИП могут наблюдаться отдельные клинические симптомы или серологические маркеры, характерные для аутоиммунной патологии, но не позволяющие установить диагноз какого-либо СЗСТ в связи с отсутствием всех необходимых критериев [6-9]. Ранее исследователи предлагали различные критерии и термины для описания вышеуказанной когорты пациентов [6-8], в связи с чем длительное время отсутствовала возможность проведения мас штабных исследований, посвященных диагностике и лечению данной патологии.
В 2015 году экспертами Европейского респираторного общества (ERS) и Американского торакального общества (ATS) был предложен термин "интерстициальная пневмонии с аутоиммунными признаками (ИПАП)" [10]. Этим термином обозначают ИИП, сопровождающуюся клиническими, серологическими и/или морфологическими признаками, которые указывают на наличие системного аутоиммунного процесса, но не соответствуют современным критериям конкретного СЗСТ. Следует отметить, что это понятие было введено группой пульмонологов и ревматологов, в первую очередь, для удобства диагностики и унификации критериев включения пациентов в клинические исследовании, однако оно не может служить основанием для выбора метода лечения.
Критерии ИПАП
Классификационные критерии ИПАП распределены на три группы, или домена – клинический, серологический и морфологический (табл. 1) [10]. Для установления диагноза ИПАП 2 необходимы наличие ИП по данным компьютерной томографии органов грудной клетки высокого разрешения (КТВР) и/или хирургической биопсии легкого и тщательное клиническое обследование для исключения известных причин ИП, в том числе СЗСТ. Кроме того, диагноз ИПАП предполагает наличие соответствующих классификационных критериев, относящихся по крайней мере к двум из указанных групп.
| 1. | Наличие интерстициальной пневмонии (по данным КТВР или хирургической биопсии легкого) + |
| 2. | Исключение альтернативных причин ИП + |
| 3. | Несоответствие критериям конкретного СЗСТ + |
| 4. | Наличие по крайней мере одного критерия из 2 или более групп: А) Клинической Б) Серологической В) Морфологической |
| А. | Клиническая группа: |
| 1) | Трещины на коже дистальных фаланг пальцев ("рука механика") |
| 2) | Кожные язвы дистальных фаланг пальцев |
| 3) | Артриты или скованность в утренние часы ≥60 мин с поражением нескольких суставов |
| 4) | Телеангиэктазии на коже ладоней |
| 5) | Феномен Рейно |
| 6) | Беспричинный отек пальцев |
| 7) | Беспричинная стойкая сыпь на коже разгибательной поверхности пальцев (симптом Готтрона) |
| Б. | Серологическая группа: |
| 1. |
Антинуклеарные антитела (АНА) в титре ⩾1:320 при наличии диффузного, гранулярного или гомогенного паттерна свечения или a) АНА при наличии нуклеолярного паттерна (в любом титре) или б) АНА при наличии центромерного паттерна (в любом титре) |
| 2. | Ревматоидный фактор в титре ⩾2N |
| 3. | Антитела к циклическому цитруллинированному пептиду (АЦЦП) |
| 4. | Антитела к двуспиральной ДНК (анти-дсДНК) |
| 5. | Анти-Ro (SS-A) |
| 6. | Анти-La (SS-B) |
| 7. | Антитела к рибонуклеопротеину |
| 8. | Анти-Smith |
| 9. | Антитела к топоизомеразе (Scl-70) |
| 10. | Антитела к тРНК-синтетазе (Jo-1, PL-7, PL-12 и другие, в том числе EJ, OJ, KS, Zo, tRS) |
| 11. | Анти-PM-Scl |
| 12. | Анти-MDA-5 |
| С. | Морфологическая группа: |
| 1. |
Предполагаемый паттерн по данным КТВР: a) Неспецифическая интерстициальная пневмония (НСИП) б) Организующая пневмония (ОП) в) Перекрест НСИП и ОП г) Лимфоцитарная интерстициальная пневмония (ЛИП) |
| 2. |
Гистологический паттерн по данным биопсии легкого: a) НСИП б) ОП в) Перекрест НСИП и ОП г) ЛИП д) Интерстициальные лимфоидные узелки с герминативными центрами |
| 3. |
Внепаренхиматозные легочные и внелегочные проявления (в сочетании с интерстициальной пневмонией) a) Необъяснимый плевральный выпот или утолщение плевры б) Необъяснимый перикардиальный выпот или утолщение перикарда в) Необъяснимое заболевание дыхательных путей (по данным легочных функциональных тестов визуализационных методов или биопсии) г) Необъяснимая легочная васкулопатия |
В клиническую группу критериев включены симптомы, относительно специфичные для ряда СЗСТ, такие как феномен Рейно, телеангиэктазии кожи ладоней, язвы дистальных фаланг пальцев [11-12]. Тем не менее, сами по себе они не позволяют установить диагноз СЗСТ. В то же время алопеция, фотодерматит, язвы слизистой оболочки полости рта, снижение массы тела, сухой синдром, изолированные миалгия и артралгия не были включены в перечень признаков ИПАП в связи с низкой специфичностью.
К серологической группе критериев отнесены аутоантитела, ассоциированных с системными аутоиммунными заболеваниями (табл. 1). Менее специфичные серологические маркеры, такие как антинуклеарные антитела (АНА) и ревматоидный фактор (РФ) в низком титре, СОЭ, С-реактивный белок (СРБ), креатинфосфаткиназа (КФК) не рассматриваются в качестве серологических критериев ИПАП [10]. Интересно, что антинейтрофильные цитоплазматические антитеал (АНЦА) не были включены в серологическую группу критериев ИПАП, так как, по мнению экспертов, они "ассоциированы с васкулитами, а не системными заболеваниями соединительной ткани". С такой точкой зрения согласиться трудно, так как ИП (особенно при наличии паттерна обычной интерстициальной пневмонии), сопровождающаяся АНЦА, чаще к миелопероксидазе, может быть первым и единственным проявлением микроскопического полиангиита, предшествующим появлению развернутой клинической картины системного васкулита [13-14].
Морфологическая группа критериев ИПАП включает в себя такие рентгенологические паттерны ИП, как неспецифическая интерстициальная пневмония (НСИП), организующая пневмония (ОП), перекрест НСИП и ОП, лимфоцитарная пневмония (ЛИП). Данные варианты легочного поражения достаточно часто встречаются при ИП, ассоциированных с СЗСТ (СЗСТ-ИП) [15-16]. Обычная интерстициальная пневмония (ОИП) (рис. 2) также может быть выявлены у пациентов с СЗСТ, хотя и встречается реже [17], поэтому она не является критерием исключения ИПАП. Тем не менее, в отличие от НСИП, ОП и ЛИП, картина ОИП не входит в перечень морфологических признаков ИПАП.
Результаты клинических исследований
С момента введения критериев ИПАП в 2015 г. были опубликованы результаты ряда клинических исследований, в основном ретроспективных, в которых изучались проявления, течение и исходы этого заболевания. Хотя основной целью разработки классификационных критериев ИПАП было выделение относительно однородной группы пациентов, тем не менее, в проведенных исследованиях выборки характеризовались значительной вариабельностью [18].
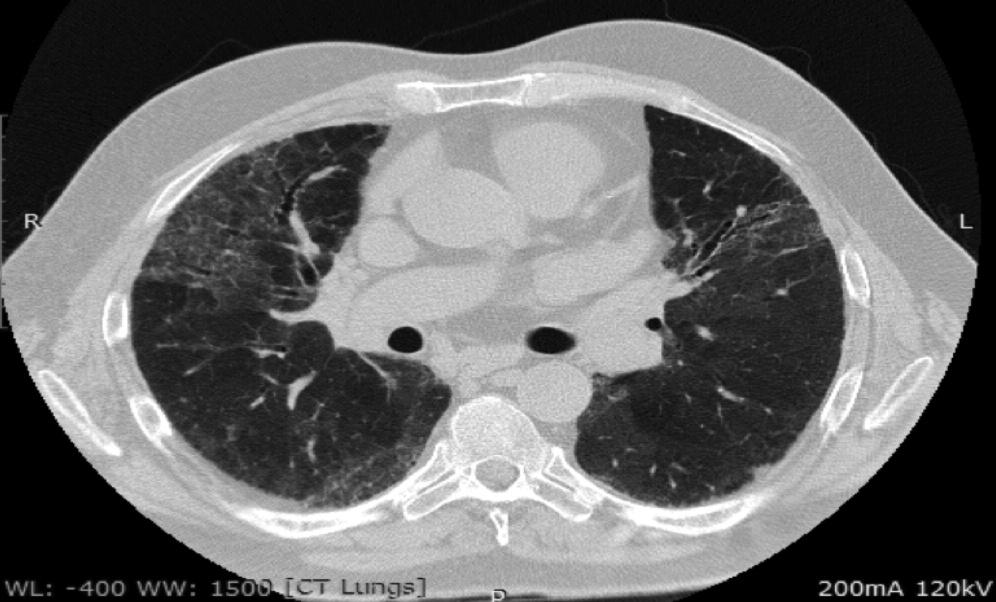
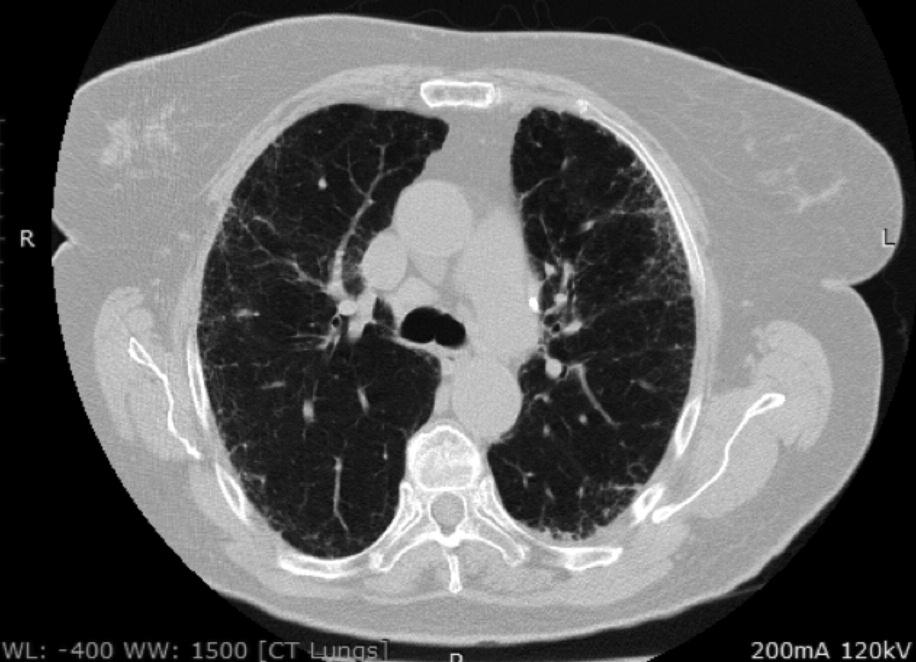
В одном из наиболее крупных исследований J. Oldham с соавт. описали группу из 144 пациентов с ИПАП: средний возраст больных составил 63 года, а приблизительно у половины из них основным рентгенологическим и/или гистологическим паттерном была ОИП [19]. У 14,6% пациентов имелись клинические и серологические критерии диагноза, у 8,3% – клинические и морфологические, у 26,4% – все три критерия. У пациентов с ИПАП и идиопатическим легочным фиброзом (ИЛФ) смертность оказалась сходной. Однако при дополнительном анализе результатов исследования было показано, что выживаемость больных ИПАП зависела от рентгенологического варианта поражения легких. При наличии картины ОИП прогноз был сходным с таковым больных с ИЛФ, тогда как у пациентов с другими рентгенологическими паттернами выживаемость была значительно выше и сопоставимой с таковой при ИП, ассоциирующейся с СЗСТ. По данным многофакторного анализа, независимыми неблагоприятными прогностическими факторами были возраст и снижение диффузионной способности легких для монооксида углерода.
В другом исследовании S. Chartrand и соавт. описали 56 пациентов с ИПАП в возрасте в среднем 54,6 года, большинство из которых были некурящими [20]. Наиболее частым паттерном ИП при КТВР и биопсии легкого была НСИП (51,8%), в то время как ОИП была выявлена всего у 9% пациентов. Более чем у половины пациентов имелись классификационные критерии, относящиеся ко всем трем группам, у 37,5% – серологические и морфологические, у 9% – клинические и морфологические, у 1,5% (1 пациент) – серологические и клинические критерии. Наиболее частым клиническими симптомами были феномен Рейно (39,3%), "рука механика" (28,6%) и симптом Готтрона (17,9%). Среди серологических критериев чаще всего встречались АНА в диагностическом титре (48,2%), анти-Ro/SS-A (42,9%), а также анти-тРНК-синтетазные антитела (АТСА) (35,7%). В исследуемой группе не отмечено ни одного летального исхода в течение периода наблюдения (284±141,3 недель).
К. Ahmad и соавт. наблюдали 57 больных с ИПАП, среди которых у 11,1% выявлены клинические и серологические классификационные критерии, у 7% – клинические и морфологические критерии, у 52,7% – серологические и морфологические критерии, у 29,2% – все три критерия [21]. Средний возраст пациентов с ИПАП составил 64,4±14 лет. Соотношение мужчин и женщин было приблизительно одинаковым. 34% пациентов курили. Наиболее частыми рентгенологическими паттернами были НСИП (42,1%) и ОИП (28%), тогда как основным внепаренхиматозным проявлением среди морфологических критериев оказалась легочная васкулопатия (17,5%). В течение периода наблюдения длительностью 16 месяцев умерли 7 пациентов. Много факторный анализ показал, что только курение было неблагоприятным прогностическим фактором. В то же время следует отметить, что в отличие от исследования J. Oldham и соавт., авторы не выявили различия выживаемости между группами пациентов с паттернами ОИП и НСИП.
В исследование Y. Ito и соавт. были включены 99 пациентов с ИПАП [22]. Один пациент с паттерном ОИП был исключен из анализа. Среди остальных 98 пациентов у 64,3% выявлен морфологический паттерн НСИП, у 20,4% – ОП, у 15,3% – перекрест НСИП и ОП. Пятилетняя выживаемость составила 71,1%, средняя продолжительность жизни после установления диагноза – 12,5 лет. Неблагоприятным прогностическим фактором было наличие паттерна НСИП, в то время как серологических предикторов прогноза выявлено не было. У 12 (12,2%) пациентов с ИПАП в динамике отмечено развитие СЗСТ (в большинстве случаев – ревматоидного артрита).
К. Yoshimura с соавт. выделили подгруппу пациентов с ИПАП среди больных с фибротическими вариантами хронической ИП (ОИП и фиброзным вариантом НСИП) [23]. Среди классификационных критериев ИПАП чаще других встречались морфологические (97% пациентов), реже – серологические (72%) и клинические (53%). У пациентов с ИПАП были отмечены более высокая выживаемость и более низкая обострений основного заболевания. При анализе подгрупп у пациентов с ИПАП, сочетавшейся с паттерном НСИП, выживаемость была выше, чем у больных с идиопатической НСИП. Также следует отметить, что в данном исследовании была выявлена тенденция к более высокой выживаемости пациентов с ИПАП, у которых определялся паттерн ОИП, по сравнению с таковой больных с ИЛФ.
H. Chung и соавт. изучали морфологические предикторы прогноза у пациентов с ИПАП [24]. У большинства из них (65,4%) при КТВР был выявлен паттерн типичной или возможной ОИП. По данным однофакторного анализа, достоверными рентгенологическими признаками, ассоциированными со снижением выживаемости, были ретикулярные изменения, зоны "сотового легкого", мозаичная вентиляция (при отсутствии эмфиземы), а также увеличение диаметра легочной артерии. В то же время наличие паттерна ОИП по данным КТВР, а также степень выраженности фиброзных изменений в легких не позволяли предсказать прогноз. По данным многофакторного анализа, независимыми достоверными предикторами неблагоприятного прогноза оказались только зоны "сотового легкого" и увеличение диаметра легочной артерии.
Таким образом, большинство данных, характеризующих группу пациентов с ИПАП, получены в ретроспективных одноцентровых исследованиях, что не позволяет экстраполироват их на всю популяцию таких больных. Этим также может частично объясняться выраженная неоднородность результатов вышеуказанных работ. В частности, в настоящее время остается до конца не выясненным прогностическое значение паттерна ОИП у пациентов с ИПАП: в ряде исследований прогноз в данной подгруппе пациентов не отличался от такового у пациентов ИЛФ, в то время как авторы других работ не выявили сходной закономерности. Кроме того, следует отметить, что в большинстве исследований не учитывалось влияние проводимой иммуносупрессивной терапии на прогноз пациентов с ИПАП. С одной стороны, это ограничивает прогностическое значение полученных результатов исследований, а, с другой стороны, не позволяет разработать рекомендации относительно рациональной фармакотерапии данной патологии.
Обсуждение
Действующие в настоящее время критерии ИПАП были предложены в 2015 г. группой экспертов в области пульмонологии и ревматологии с целью выделения отдельной когорты пациентов с ИИП и признаками системных аутоиммунных заболеваний, не соответствующих классификационным критериям конкретного СЗСТ. Преимущество предложенной классификации заключается в том, что она заменила множество введенных ранее и различающихся между собой определений ИПАП, а также стала основой для разработки критериев включения пациентов в клинические исследования, необходимые для изучения этой патологии.
С учетом новых данных, полученных в группах пациентов с ИПАП, ряд исследователей предлагают различные варианты пересмотра и доработки существующих критериев заболевания. В частности, G. Sambataro и соавт. [18] указывают на то, что наличие среди критериев ИПАП специфичных или даже патогномоничных признаков СЗСТ (симптом Готтрона [25], "рука механика", антитела к цитруллинированному пептиду, АТСА и ряд других аутоантител) не является рациональным. Это объясняется тем, что классификационные критерии ИПАП пересекаются с таковыми недифференцированного заболевания соединительной ткани [26-27] или с критериями, предложенными для ранней диагностики некоторых СЗСТ. Таким образом, в дебюте часть СЗСТ могут ложно классифицироваться как ИПАП. Примером могут служить критерии ранней диагностики системной склеродермии (very early diagnosis of systemic sclerosis, VEDOSS) [28], которые позволяют идентифицировать пациентов с высоким риском развития этого заболевания. Данная группа пациентов представляет большой интерес, особенно учитывая тот факт, что наличие явных "склеродермических" признаков позволяет не только установить диагноз, но и начать лечение на ранней стадии заболевания.
Кроме того, критериям ИПАП может соответствовать часть пациентов с антисинтетазным синдромом. Принимая во внимание отсутствие единых международных критериев этого синдрома, пациентам с ИП и антисинтетазными антителми (АТСА) может был установлен диагноз как антисинтетазного синдрома, так и ИПАП, несмотря на высокий риск развития у них остальных клинических проявлений классической триады симптомов (артрит и/или миозит) [29]. В отношении антисинтетазного синдрома существует еще одна проблема: с одной стороны, у всех пациентов с ИП необходимо определять антитела, ассоциированные с воспалительными миопатиями, однако на практике в большинстве клинических лабораторий полный перечень данных аутоантител рутинно не исследуется, поэтому антисинтетазный синдром может остаться недиагностированным.
Рядом исследователей были предложены дополнения к существующему определению ИПАП, в частности в серологическую группу критериев рекомендовано включение дополнительных антител, таких как АНЦА [30] и анти-Ku антитела [31]. Уже описаны когорты пациентов с АНЦА-позитивной ИП. Тем не менее, на настоящий момент количество данных относительно этой группы больных весьма ограничено. H. Yamada описал группу из 92 пациентов с ИП, которую ранее классифицировали как ИЛФ с наличием АНЦА [32]. В данной когорте у 35,8% отмечалось наличие АНЦАассоциированного васкулита в дебюте заболевания, еще у 17,4% пациентов он развился в течение нескольких лет после установления диагноза, а у оставшихся 46,8% пациентов ИП оставалась единственным клиническим проявлением заболевания. У части пациентов при биопсии легкого определялись признаки васкулита с поражением интерстиция и/или бронхов. Авторами статьи предложено два возможных механизма формирования интерстициальных изменений в легких при наличии АНЦА. Первый предполагает развитие интерстициального фиброза в результате субклинических альвеолярных геморрагий. В соответствии со второй гипотезой в ответ на образование воспалительных цитокинов миелопероксидаза экспрессируется на поверхности нейтрофилов, что может приводить к фиксации циркулирующих АНЦА с последующей дегрануляцией нейтрофилов и высвобождением активных форм кислорода, которые вызывают повреждение легочной ткани и развитию фиброза. Второй механизм, в частности, способен объяснить развитие АНЦА-ассоциированной ИП без признаков системного васкулита.
Таким образом, текущие диагностические критерии ИПАП охватывают весьма неоднородную группу пациентов, в связи с чем в большинстве исследований проводился поиск прогностических факторов выживаемости пациентов с ИПАП. Особый интерес вызывает морфологический паттерн ОИП, относительно значения которого в настоящее время нет единого мнения. Отсутствие ОИП в группе морфологических критериев ИПАП, с одной стороны, и более низкая выживаемость в данной подгруппе пациентов по результатам ряда исследований, с другой стороны, ставят вопрос о том, не должны ли пациенты с ОИП исключаться из клас сификационной группы ИПАП. Тем не менее, до появления данных проспективных исследований, подтверждающих внутригрупповую неоднородность ИПАП, выявления достоверных предикторов прогноза выживаемости и ответа на различные варианты терапии, пациентов с ОИП следует включать в группу ИПАП при наличии клинических и/или серологических критериев данного диагноза [33].
В ближайшем будущем необходимо проведение проспективных клинических исследований с целью изучения эффективности иммуносупрессивных, и возможно, антифибротических препаратов у пациентов с ИПАП. При большинстве вариантов СЗСТ-ИП и не-ОИП вариантах идиопатической ИП основной медикаментозной терапии являются глюкокортикостероиды, которые в части случаев сочетают с иммунодепрессивными и генно-инженерными биологическими препаратами, однако они неэффективны при ИЛФ, который представляет собой вариант идиопатической ИП с рентгенологическим паттерном ОИП [34,35]. Учитывая доказанную эффективность антифибротической терапии у пациентов с ИЛФ, не исключена возможность ее применения у части пациентов с ИПАП.
Заключение
Диагноз ИПАП был введен с целью унификации группы пациентов с ИП, у которых определяются отдельные симптомы системных аутоиммунных заболеваний. Тем не менее, данные клинических исследований указывают на то, что текущее определение ИПАП, вероятно, не позволяет выделить однородную популяцию больных. Уточнение классификационных критериев ИПАП может оказаться полезным для выделения отдельных вариантов этого состояния, отличающихся по клиническому течению, ответу на терапию и прогнозу, в том числе вероятности развития определенных системных заболеваний соединительной ткани. Для подтверждения данной гипотезы требуется проведение многоцентровых проспективных исследований с междисциплинарным подходом к диагностике и выбору терапии пациентов с ИПАП.
Используемые источники
- Travis WD, Costabel U, Hansell DM, et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2013:188(6):733–48.
- Fischer A, du Bois R. Interstitial lung disease in connective tissue disorders. Lancet 2012;380:689–698.
- Tzelepis GE, Toya SP, Moutsopoulos HM. Occult connective tissue diseases mimicking idiopathic interstitial pneumonias. Eur Respir J 2008;31:11–20.
- Mittoo S, Gelber AC, Christopher-Stine L, et al. Ascertainment of collagen vas- cular disease in patients presenting with interstitial lung disease. Respir Med 2009;103:1152–58.
- Castelino FV, Varga J. Interstitial lung disease in connective tissue diseases: evolv- ing concepts of pathogenesis and management. Arthritis Res Ther 2010;12:213.
- Kinder BW, Collard HR, Koth L, et al. Idiopathic nonspecific interstitial pneu- monia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med 2007;176:691–7.
- Fischer A, West SG, Swigris JJ, et al. Connective tissue disease-associated inter- stitial lung disease: a call for clarification. Chest 2010;138:251–6.
- Vij R, Noth I, Strek ME. Autoimmune-featured interstitial lung disease: a distinct entity. Chest 2011;140:1292–9.
- Corte TJ, Copley SJ, Desai SR, et al. Significance of connective tissue disease fea- tures in idiopathic interstitial pneumonia. Eur Respir J 2012;39:661–8.
- Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015;46:976–87.
- LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): clas- sification, subsets and pathogenesis. J Rheumatol 1988;15:202–5.
- LeRoy EC, Medsger TA. Criteria for the classification of early systemic sclerosis. J Rheumatol 2001;28:1573–76.
- Ando M, Miyazaki E, Ishii T, et al. Incidence of myeloperoxidase anti-neutrophil cytoplasmic antibody positivity and microscopic polyangitis in the course of idio- pathic pulmonary fibrosis. Respir Med 2013;107:608–15.
- Tzelepis GE, Kokosi M, Tzioufas A, et al. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J 2010;36:116–21.
- Tanaka N, Newell JD, Brown KK, et al. Collagen vascular disease-related lung disease: high-resolution computed tomography findings based on the pathologic classification. J Comput Assist Tomogr 2004;28:351–60.
- Hwang JH, Misumi S, Sahin H, et al. Computed tomographic features of idio- pathic fibrosing interstitial pneumonia: comparison with pulmonary fibrosis relat- ed to collagen vascular disease. J Comput Assist Tomogr 2009;33:410–5.
- Kim EJ, Elicker BM, Maldonado F, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2010;35:1322–8.
- Sambataro G, Sambataro D, Torrisi SE. State of the art in interstitial pneumonia with autoimmune features: a systematic review on retrospective studies and sug- gestions for further advances. Eur Respir Rev 2018;27:170139.
- Oldham JM, Adengunsoye A, Valenzi E, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J 2016;47:1767–75.
- Chartrand S, Swigris JJ, Stanchev L, et al. Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir Med 2016;119:150-4.
- Ahmad K, Barba T, Gamondes D, et al. Interstitial pneumonia with autoimmune features: clinical, radiologic and histological characteristics and outcome in a series of 57 patients. Respir Med 2017;123:56–62.
- Ito Y, Arita M, Kumagai S, et al. Serological and morphological prognostic fac- tors in patients with interstitial pneumonia with autoimmune features. BMC Pulm Med 2017;17:111.
- Yoshimura K, Kono M, Enomoto Y, et al. Distinctive characteristics and prog- nostic significance ofinterstitial pneumonia with autoimmune features in patients with chronic fibrosing interstitial pneumonia. Respir Med 2018;137:167-75.
- Chung JH, Montner SM, Adegunsoye A. CT findings, radiologic-pathologic cor- relation, and imaging predictors of survival for patients with interstitial pneumonia with autoimmune features. Am J Roentgenol 2017;208(6):1229-36.
- Kholer RA, Montemarano A. Dermatomyositis. Am Fam Physician 2001;64:1565–72.
- Mosca M, Neri R, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): a review of the literature and a proposal for preliminary classification criteria. Clin Exp Rheumatol 1999;17:615–20.
- Doria A, Mosca M, Gambari PF, et al. Defining unclassifiable connective tissue diseases: incomplete, undifferentiated or both? J Rheumatol 2005;32:213–5.
- Matucci-Cerinic M, Allanore Y, Czirják L, et al. The challenge of early systemic sclerosis for the EULAR Scleroderma Trial and Research Group (EUSTAR) community. It is time to cut the Gordian knot and develop a prevention or rescue strategy. Ann Rheum Dis 2009;68:1377–80.
- Scirè CA, Gonzalez-Gay MA, Selva-O’Callaghan A, et al. Clinical spectrum time course of interstitial pneumonia with autoimmune features in patients positive for antisynthetase antibodies. Respir Med 2017;132:265–6.
- Alba MA, Flores-Suárez LF, Henderson AG, et al. Interstitial lung disease in ANCA vasculitis. Autoimmun Rev 2017;16:722–9.
- Ghirardello A, Borella E, Beggio M, et al. Myositis autoantibodies and clinical phenotypes. Auto Immun Highlights 2014;5:69–75.
- Yamada H. ANCA: associated lung fibrosis. Semin Respir Crit Care Med 2011;32:322–7.
- Brovko M, Akulkina L, Sholomova V, et al. Usual interstitial pneumonia: A dis- tinct group within interstitial pneumonia with autoimmune features? Respirology 2018;23(10): 58.
- Авдеев С.Н. Идиопатические интерстициальные пневмонии: особенности клинической картины и лечения. Фарматека 2009;19:12-9 [Avdeev SN. Idiopathyic interstitial pneumonia: clinical picture and treatment. Farmateka 2009;19:12-9 (In Russ.)].
- Аверьянов А.В., Лесняк В.Н., Коган Е.А. Редкие заболевания легких: диаг- ностика и лечение (под ред. А.В. Аверьянова) М., МИА, 2016, 248 с.