Проблемы диагностики и лечения локального AL-амилоидоза
Описать клинические проявления, подходы к диагностике и лечению локального амилоидоза AL-амилоидоза (ALL ).
Были обследованы 30 больных ALL , у которых наблюдалось изолированное типичное поражение одного органа при отсутствии моноклональной гаммапатии. Группу сравнения составили 110 пациентов с системным AL-амилоидозом. При формулировании принципов дифференциального диагноза применяли многофакторный метод анализа соответствий.
Самыми частыми вариантами ALL были ларинготрахеобронхиальная форма (n=12) и поражение конъюнктивы или ткани глазницы (n=8), реже встречалось поражение легких (n=3), кожи (n=3), мочевого пузыря (n=2), головного мозга (n=1) и мягких тканей (n=1). Депозиты амилоида могли быть опухолевидными (n=17) или относительно диффузно инфильтрировали ткань (n=13). Cреднее время, необходимое для установления диагноза, составило 23 мес (8-55 мес). Для дифференциальной диагностики с системным AL-амилоидозом необходимо исключить наличие протеинурии и моноклональной гаммапатии с помощью высокочувствительного иммунохимического исследования и иммунофенотипирования костного мозга. После удаления амилоидных депозитов у каждого четвертого пациента отмечался рецидив ALL .
Для лечении ALL в большинстве случаев достаточно малотравматичного иссечения тканей, однако высокий риск рецидивирования требует поиска других методов лечения, таких как лучевая терапия с целью уничтожения локального амилоидогенного клона плазматических клеток.
Амилоидоз – это большая группа заболеваний, которые характеризуются отложением в тканях фибриллярного гликопротеида со специфическим свойством двойного лучепреломления. При микроскопии препаратов амилоида, окрашенных конго красным, в поляризованном свете цвет фибрилл изменяется с красного на яблочно-зеленый. Это свойство обусловлено бета-складчатой конформационной струк турой, которая приобретена амилоидом благодаря ее наличию в основном белкепредшественнике. Многообразие таких белков, а их известно более 30, и определяет многочисленность форм амилоидоза [1].
Одним из традиционных принципов классификации амилоидоза является выделение локальных и системных форм амилоидоза. Уже в первой клинической классификации амилоидоза, предложенной в 1935 г. H. Rei mann и соавт., наряду с первичным, вторичным и ассоциированным с множественной миеломой формами системного амилоидоза, был выделен опухолевидный амилоидоз, т.е. локальный вариант заболевания [2]. Соглас но этой классификации, изолированное поражение одного органа позволяет диагностировать локальный амилоидоз, а поражение нескольких органов указывает на системный вариант заболевания.
На первый взгляд этот подход к классификации является слишком механистичным, тем не менее, исследователи безошибочно почувствовали принципиальную патогенетическую разницу между локальными и системными формами амилоидоза. Так, локальный амилоидоз, по-видимому, не обладает способностью к трансформации в системный амилоидоз с прогрессирующим поражением внутренних органов и обычно не приводит к гибели пациентов; эта форма проявляется только нарушением функции пораженного органа [3,4]. H. Reimann и соавт. в своей классификации акцентировали внимание на опухолевидности локальных форм амилоидоза.
В связи с изолированностью патологического процесса пациенты с локальным амилоидозом часто обращаются к специалистам узкого профиля (оториноларингологу, оф таль мологу, урологу, нейрохирургу и др.), которые хирургическим методом удаляют амилоидные отложения. Однако опыт наблюдения за больными локальным амилоидозом показывает, что однократное удаление амилоидных образований нередко является неэффективным, болезнь может рецидивировать, а повторные хирургические вмешательства создают угрозу инвалидизации больных. Имеются также диагностические трудности, так как системные формы амилоидоза могут длительно проявляться преимущественным поражением одного органа.
Принципиальное отличие между системными и локальными формами амилоидоза заключается в том, что при системном амилоидозе белок-предшественник, запускающий сборку амилоида, циркулирует в крови и поэтому способен поражать любой орган. Даже при отсутствии клинических признаков поражения какоголибо органа его биопсия, как правило, позволяет выявить амилоид. При локальных формах синтез белкапредшественника отмечается локально, только в месте депозиции амилоида, при этом белок-предшественник не способен проникнуть в кровоток.
Наиболее многочисленной является группа локального церебрального амилоидоза – депозиция β-белка (Аβ-амилоидоз) приводит к болезни Альцгеймера и синдрому Дауна (синтез β-белка кодируется 21 хромосомой, а трисомия этой хромосомы приводит к гиперпродукции этого белка и его отложению в виде амилоида в головном мозге) [5], депозиция прионового белка является причиной губкообразных энцефалопатий (куру, болезнь Крейтцфельда-Якоба, фатальная семейная бессонница, синдром Гертсманна-Штраус слера-Шейнкера), известны наследственные формы амилоидной ангиопатии мозговых сосудов с развитием кровоизлияний (ACys-амилоидоз – цистатиновый, ABri-амилоидоз) [6]. Самой частой формой локального амилоидоза является амилоидоз пред сердий, обусловленный отложением предсердного натрийуретичес кого фактора (AANF-амилоидоз) [7]. Известна также ятрогенная форма локального амилоидоза, вызванного отложением инсулина в местах введения препарата больным сахарным диабетом [8,9].
Перечисленные формы локального амилоидоза могут поражать только ткани, в которых синтезируются соответствующие белки-предшественники. Так, не может быть Aβ- или прионового амилоидоза с поражением гортани или мочевого пузыря, так же как депозиции в этих органах предсердного натрийуретического фактора. Среди локальных форм в этих органах может отмечаться только секреция амилоидогенных легких цепей иммуноглобулинов (AL-амилоидоз) поселившимся в ткани клоном плазматических клеток. Таким образом, при локальных формах амилоидоза представляется возможным проведение дифференциального диагноза по клиническим особенностям заболевания, в первую очередь, с учетом органной локализации амилоидных депозитов. При системных формах амилоидоза дифференцирование по клиническим признакам без объяснения патогенеза представляется крайне затруднительным. В то же время, следует помнить, что среди системных форм амилоидоза самым распространенным также является AL-амилоидоз, при котором амилоидогенный клон плазматических клеток в отличие от локального варианта поселяется в костном мозге, структура и функция которого приспособлены для системной продукции иммуноглобулинов, и аномальные иммуно глобулины поступают в системный кровоток. Следо вательно, дифференциальный диагноз ALL предполагает учет патогенетических различий с системным вариантом и, таким образом, умение отличить костномозговой клон плазмацитов от локального.
Целью исследования было описать клинические проявления, подходы к диагностике и лечению ALL.
Материал и методы
В исследование включали всех пациентов с ALL. Диагноз амилоидоза устанавливался на основании выявления амилоида в биоптате пораженного органа при окраске конго красным при наличии характерного желто-зеленого свечения в поляризованном свете. Обязательным условием для подтверждения ALL у пациентов с типичным поражением одного органа считали отсутствие моноклональной гаммапатии по результатам электрофоретического исследования сыворотки крови и мочи с применением высокочувствительных методов иммунофиксации и количественного определения свободных легких цепей иммуноглобулинов методом Freelitе. При наличии потенциального риска иных биохимических вариантов амилоидных депозитов (при поражении головного мозга) или противоречивых результатах определения моноклональной гаммапатии AL-тип амилоида подтверждали по результатам иммуногистохимического определения в составе амилоида моноклональных легких цепей иммуноглобулинов лямбда или каппа типа. Всем пациентам проводили стандартные лабораторные и инструментальные исследования для выявления поражения различных органов и систем (включая ультразвуковое почек и эхокардиографию), при необходимости использовали функциональные (спирометрия), эндоскопические (ларингоскопия, бронхоскопия, цистоскопия) и лучевые (компьютерная томография органов грудной клетки и головного мозга, магнитно-резонансная томография головного мозга, позитронно-эмиссионная томография/компьютерная томография) методы исследования.
Статистическая обработка результатов проводилась преимущественно непараметрическими методами (Z-критерий, критерий Манна-Уитни), при формулировании принципов дифференциального диагноза применяли многофакторный метод анализа соответствий. Анализ данных проводился с применением программного обеспечения Statistica 7.0.
Результаты
ALL был диагностирован у 30 (9%) из 343 пациентов с амилоидозом (n=343), которые находились на амбулаторном или стационарном лечении в клинике им. Е.М. Тареева с 1995 по 2018 г. Доля ALL среди других форм амилоидоза была приблизительно равной таковой ATTR (8%), однако ALL встречался почти в 5 раз реже системного AL (41%) и АА-амилоидоза (41%). Медиана возраста больных ALL составила 47 лет (межквартильный размах 36-55 лет). Самой молодой была 4-летняя пациентка с поражением гортани. Еще у 3 больных молодого возраста (18, 22 и 24 лет) наблюдался амилоидоз кожи. Среди пациентов с ALL преобладали женщины (77%).
Самым частым вариантом ALL была ларинготрахеобронхиальная форма (n=12), которая в сочетании с ALL легких (n=3), была диагностирована у половины больных. Еще у трети пациентов отмечалось отложение амилоида в конъюнктиве или ткани глазницы (n=8). Реже встречался амилоидоз кожи (n=3), мочевого пузыря (n=2), головного мозга (n=1) и мягких тканей (n=1).
У всех 12 пациентов с амилоидозом верхних дыхательных путей наблюдалось поражение гортани, проявлявшееся осиплостью голоса, а также затруднением дыхания (у 9) в результате сужения дыхательных путей депозитами амилоида. У 2 больных имелась выраженная бронхиальная обструкция, не уменьшавшаяся под действием ингаляций b2-адреномиметиков или м-холинолитиков. У 1 пациента, несмотря на неоднократное хирургическое удаление амилоидных депозитов, амилоидоз рецидивировал, а его распространение привело к тяжелой прогрессирующей дыхательной недостаточности, осложнившейся легочной инфекцией и смертью больного. У 3 больных легочным ALL заболевание протекало бессимптомно. ALL мочевого пузыря проявлялся безболевой рецидивирующей макрогематурией и учащением мочеиспускания. При цистоскопии у этих 2 пациентов определялось диффузное утолщение стенки мочевого пузыря, заставляющее подозревать злокачественное новообразование, однако при трансуретральной биопсии мочевого пузыря был диагностирован ALL. Амилоидоз кожи характеризовался упорным кожным зудом, который заставлял пациентов прибегать к специальным приспособлениям для расчесывания кожи и вызывал социальную дезадаптацию. При отложении амилоида в конъюнктиве и мягких тканях глаза или мягких тканях другой локализации пациенты обычно обнаруживали опухолевидные депозиты самостоятельно и незамедлительно обращалось к врачу, предполагая онкологическое заболевание. Депозиты, как правило, были безболезненными, на их поверхности могли определяться кровоизлияния, а в толще просвечивали сосуды. У некоторых больных амилоидоз конъюнктивы сопровождался отеком век, а у одного наблюдалось опущение верхнего века, что существенно нарушало зрительную функцию.
Депозиты амилоида имели опухолевидную форму (у 17) или относительно диффузно инфильтрировали пораженную ткань (у 13). У всех 3 больных с поражением кожи отмечалась диффузная инфильтрация. В остальных случаях затруднительно обсуждать преимущественный характер отложения амилоида из-за малочисленности подгрупп. У 12 из 17 больных с опухолевидным амилоидозом предполагали неопластический процесс, а у остальных 5 больных обсуждались воспалительные заболевания соответствующей локализации (хронический обычный или гипертрофический ларингит, лимфаденит, туберкулез легких). У 5 из 13 больных с диффузным отложением амилоида также первоначально предполагали наличие опухоли. Медиана времени, в течение которого была проведена биопсия пораженной ткани, составила 1 месяц, однако межквартильный диапазон был достаточно большим (033 месяца), а у 11 (37%) больных биопсия была проведена по разным причинам только через 1-23 года. Отсрочки в биопсии более года не были связаны с преимущественно инфильтративным характером депозиции амилоида или наличием предположения об опухоли. Наибольшие отсрочки в проведении биопсии отмечались у больных с ларинготрахеальным ALL (медиана 25 мес, межквартильный размах 0-50 мес) и поражением кожи (медиана 60 мес, межквартильный размах 13-134 мес). У всех 3 больных с поражением легких биопсию провели в течение первых 2 мес от момента обращения к врачу, а диагноз был установлен через 4, 11 и 13 мес от начала заболевания. После биопсии диагноз амилоидоза в целом по группе устанавливали в среднем спустя 6 мес (межквартильный диапазон 0-40 мес) и только у половины больных в течение первого года после биопсии. Быстрее всего биопсию проводили больным с локальным амилоидозом конъюнктивы или других сред глаза (медиана 4 мес, межквартильный размах 1-6 мес), а диагноз амилоидоза устанавливали в среднем через 6 месяцев (1-20 мес) после начала заболевания. В целом медиана времени, необходимого для установления диагноза амилоидоза, составила 23 мес (межквартильный диапазон 8-55 мес).
ALL резко отличается по прогнозу от системного варианта. При естественном течении системного AL средняя продолжительность жизни составляет 12 мес [10]. Современные химиотерапевтические методы лечения, направленные на уничтожение амилоидогенного клона плазматических клеток, позволили значительно улучшить показатели выживаемости. Так, по нашим данным медиана выживаемости больных системным AL в настоящее время увеличилась до 5 лет, а среди достигших полной молекулярной ремиссии – 7 лет [11]. При ALL причины смерти, как правило, не связаны с амилоидозом. Как уже указывалось, только у одного пациента поражение амилоидом респираторного тракта легких привело к прогрессированию дыхательной недостаточности и летальному исходу.
Для анализа подходов к дифференциальной диагностике ALL мы использовали контрольную группу. Ее составили 110 пациентов с системным AL-амилоидозом, которые в течение длительного времени наблюдаются в клинике. У 56 (51%) из них наблюдался “ограниченный" вариант амилоидоза, который характеризовался длительным сохранением поражения только одного органа в начале заболевания. Длительность преимущественно моноорганного течения заболевания составила 17 мес (межквартильный диапазон 11-32 мес), т.е. существенно превышала период естественной выживаемости больных системным AL. В этой связи ограниченный вариант течения AL создает угрозу поздней диагностики системного AL и, соответственно, несвоевременного лечения. Риск ошибочного диагноза был особенно велик у 10 больных, у которых первыми симптомами были поражение кожи или мягких тканей, периорбитальная геморрагия, макрогематурия или осиплость голоса. У части больных наблюдались системные проявления, однако они были мало выражены и могли быть выявлены только при специальном обследовании. В то же время при преимущественном поражении сердца, почек или нервной системы диагноз ALL мало вероятен, так как поражение этих органов для него не характерно. Следует отметить, что в литературе имеются описания локальной AL-амилоидомы кишечника, поэтому можно предположить возможность локального поражения и других внутренних органов [12].
С помощью многофакторного метода соответствий мы изучили предсказательное значение следующих признаков в дифференциальной диагностике локального и системного AL-амилоидоза: 1) протеинурия более 0,5 г/л или продвинутая стадия (3-5) хронической болезни почек; 2) клинические, электрокардиографические и/или эхокардиографические признаки поражения сердца; 3) утолщение задней стенки левого желудочка и/или межжелудочковой перегородки >12 мм (наиболее характерный для системного амилоидоза с поражением сердца признак); 4) поражение нервной системы (ортостатическая гипотензия, моторная диарея, периферическая полиневропатия); 5) поражение кожи, мягких тканей, периорбитальные геморрагии, макрогематурия или осиплость голоса, характерные для ALL; 6) возраст начала заболевания; 7) моноклональная гаммапатия.
Предполагалось, что моноклональная гаммапатия обладает доминирующим значением в дифференциальной диагностике, поскольку непосредственно указывает на принципиальное патогенетическое различие между локальным и системным амилоидозом. По этой причине на первом этапе многофакторного анализа этот параметр не был включен, чтобы избежать нивелирования значения других факторов. По нашим данным, одним из отличительных признаков ALL является более молодой возраст начала заболевания: при ALL медиана составила 47 лет (межквартильный диапазон 36-55 лет), при системном – 54 года (межквартильный диапазон 48-65 лет; р=0,000195). С точки зрения диагностической эффективности параметр возраста самостоятельного значения не имел, так как найти оптимальные значения чувствительности и специфичности не удалось. Исходя из приоритета специфичности над чувствительностью, было выбрано значение возраста 48 лет (специфичность – 71%, чувствительность – 55%).
Первый этап многофакторного анализа показал, что 59,9% всех соответствий между переменными может быть объяснено использованием только двух осей, указывающих на удаленность между параметрами по результатам χ2-сравнения таблиц частот признаков (рис. 1). Наращивание дополнительных осей несущественно повышает число объясненных соответствий, но значительно затрудняет интерпретацию результатов. Например, по третьей оси уже исчезает противопоставление между локальным и системным AL. Наиболее информативной является первая ось сравнения, которая объясняет 46,9% соответствий. Согласно этой оси точки локального и системного амилоидоза максимально удалены друг от друга, по отношению к началу оси координат (точка х=0; у=0) формируют практически прямую линию, т.е. именно по этой оси удобно оценивать взаимосвязь включенных параметров с локальным или системным AL. Точки других параметров формируют довольно четкие совокупности тяготения либо к точке локального, либо системного амилоидоза. Это в первую очередь определяется по углу, который формируют эти точки с точкой системного/локального амилоидоза по отношению к началу координат, а также по удаленности точек друг от друга. Согласно этому принципу очень четко выделяется совокупность точек вокруг системного амилоидоза, что позволяет сформулировать следующее положение – у больных с признаками протеинурического поражения почек, сердца (в первую очередь с утолщением миокарда более 12 мм), нервной системы, в возрасте старше 48 лет при отсутствии признаков, характерных для ALL, с высокой вероятностью можно предполагать системный вариант AL-амилоидоза. Совокупность локального варианта заболевания очерчена менее четко. Это особенно видно при анализе второй оси, по которой точки локального и системного амилоидоза практически не противопоставлены и при исключении первой оси формируют практически единую совокупность. Вторая ось в большей мере противопоставляет параметры возраста начала заболевания и наличия/отсутствия кардиопатии, при этом точки параметра возраста оказываются практически равноудаленными как от точки локального, так и системного амилоидоза, а углы с началом координат у обеих точек возраста являются близкими по своим значениям. Малая информативность параметра кардиопатии связана с включением в него большого количества неспецифических признаков. У многих больных ALL имелись артериальная гипертония и другие распространенные заболевания с поражением сердца, поэтому отсутствие признаков поражения сердца не увеличивало шансы наличия ALL. В этой связи при дальнейшем анализе параметры возраста начала заболевания и наличия поражения сердца в схеме дифференциального диагноза не учитывались, а в качестве аналога амилоидоза сердца оценивали только наличие утолщения миокарда. Точка наличия признаков, характерных для локального амилоидоза, несмотря на свое тяготение к точке ALL, была более удалена, чем точка отсутствия нефропатии. По-видимому, это связано с тем, что поражение кожи, глаз, мягких тканей, осиплость голоса нередко сопутствуют системному амилоидозу. Нормальная толщина миокарда и отсутствие поражения нервной системы также имели ограниченную диагностическую значимость в представленных осях анализа соответствий.
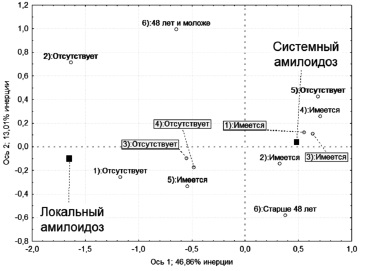
В связи с этими рассуждениями на втором этапе из анализа соответствий были исключены параметры наличия поражения сердца и возраста начала амилоидоза, но был добавлен параметр наличия моноклональной гаммапатии. Полученная диаграмма соответствий представлена на рис. 2. Анализ скорригированной системы параметров показал ее более высокую диагностическую эффективность, сумма двух основных осей позволяла объяснить 65,9% соответствий между параметрами, при этом сохранялось противопоставление точек системного и локального амилоидоза. В представленной системе координат потеряли свой смысл параметры утолщения миокарда и наличия поражения нервной системы, практически малоинформативной является также оценка клинических признаков, характерных для локального амилоидоза. Таким образом, для обоснованного предположения о ALL и исключения системного варианта достаточно исключить наличие моноклональной гаммапатии и протеинурии.
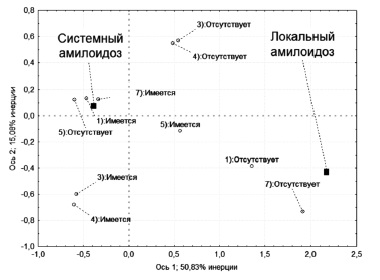
В то же время необходимо отметить, что исключение моноклональной гаммапатии с помощью иммунохимического исследования с применением высокочувствительных методов, несмотря на очень высокую информативность, не обладает абсолютной значимостью. У одного нашего пациента системным AL подтвердить наличие аномального клона плазматических клеток удалось только методом иммунофенотипирования костного мозга с применением панели плазмацитарных антигенов (CD 38, 19, 45, 56) и оценки клональности плазматических клеток по рестрикции каппа/лямбда вариантов легких цепей иммуноглобулинов. По всей видимости, очевидным доказательством локального присутствия клона плазматических клеток является также его обнаружение в биоптатах, в которых подтверждено наличие амилоида.
Лечение ALL обычно заключается в удалении выявленных амилоидных депозитов. У 18 (60%) из 30 больных применяли стандартные методы хирургического иссечения амилоида. В случае амилоидоза верхних дыхательных путей амилоид обычно удаляли эндоларингоскопическим методом (у 8), однако у 3 пациентов, в том числе у 2 после эндоскопического удаления амилоида, была также проведена вынужденная трахеостомия. При поражении легких у всех 3 пациентов была выполнена видеоторакоскопическая атипичная резекция легкого. У 2 больных с глубоким поражением мягких тканей глазницы применялась орбитотомия (по Смиту и нижняя трансконъюнктивальная эксплораторная). У пациентки с локальным амилоидозом височной доли головного мозга хирургическое удаление амилоидомы методом костнопластической трепанации осложнилось развитием гемианопсии из-за необходимости удалять обширный участок пораженной ткани мозга. Во всех случаях стандартного хирургического вмешательства имелся высокий риск инвалидизирующих анатомофизиологических изменений при необходимости повторной операции.
Менее травматичные радио- или лазерная эксцизия или же аргоно-плазменная деструкция были применены только у 5 (17%) больных, в том числе у 3 пациентов с амилоидозом конъюнктивы и 2 больных с ларинготрахеобронхиальным амилоидозом. У 3 больных из-за диффузного инфильтративного поражения кожи хирургическое лечение было трудно выполнимым, лечение ограничивалось малоэффективными способами амилоидорезорбции с помощью аппликаций димексида (у 2 больных) или симптоматической противозудной терапии. Инстилляции димексида применялись также у 1 больной с амилоидозом мочевого пузыря, что, однако, было связано с частыми цистоскопиями и риском рецидивирующей мочевой инфекции при малой эффективности димексида. У другой больной с амилоидозом мочевого пузыря тактика ведения ограничивалась наблюдением. В обоих случаях локального амилоидоза мочевого пузыря хирургическое лечение было невозможным из-за диффузного инфильтративного характера депозиции амилоида, что потребовало бы полного удаления мочевого пузыря.
У 8 (27%) из 30 больных отмечалось рецидивирование амилоидоза в среднем через 57 мес (межквартильный диапазон 18-79 мес).
Обсуждение
Результаты исследования показывают, что ALL является редким заболеванием, о частоте которого можно судить по распространенности системного AL-амилоидоза. Показано что системный AL-амилоидоз встречается у 8 человек на 1 млн населения [13,14]. Поскольку среди наших пациентов шансы развития ALL 4,7 раз меньше по сравнению с системным AL можно ожидать, что частота ALL приблизительно составляет 1-2 на 1 млн населения. К таким же результатам приводит сравнение с родственным ALL заболеванием – солитарной экстрамедуллярной плазмацитомой, при которой опухолевый клон плазматических клеток также не мигрирует в костный мозг [19], а заселяет мягкие ткани, что является одним из указаний на биологическую близость к клону, индуцирующему ALL. При этом плазмоцитома, повидимому, также может быть противопоставлена множественной миеломе, как ALL противопоставлен системному варианту. По мнению R. Liebross и соавт., доля плазмацитомы не превышает 2% среди всех плазмоклеточных дискразий [15,16], в то время как частота множественной миеломы в США составляет 4,3 на 100 тыс населения [17]. Несложный арифметический пересчет позволяет вывести данные, совпадающие с указаниями G. Dores и соавт., – частота солитарной экстрамедуллярной плазмацитомы составляет около 1 на 1 млн населения [18].
Анализ преимущественной локализации ALL показал, что несмотря на принципиальную возможность практически любой локализации, у 86% поражались ткани выше диафрагмы, что в целом также совпадает с преимущественной локализацией экстрамедуллярной плазмацитомы, которая по наблюдению Alexiou и соавт. в 85% случаев поражает область головы и шеи [16,20]. Плазмацитома отличается от множественной миеломы более молодым возрастом пациентов [18,21,22], такие же отношения мы наблюдали между ALL и системным AL-амилоидозом [11], обусловленным миграцией клона плазматических клеток в костный мозг. Наконец, неоднократное обнаружение в составе амилоида клональных плазмацитов является наиболее веским аргумментом в пользу сопоставления ALL и плазмацитомы и оправдывает сходство подходов к диагностике и лечению этих двух заболеваний.
Существенной проблемой остается длительный срок установления диагноза ALL. Сравнение более длительных сроков диагностики ларинготрахеобронхиального амилоидоза и родственного этой форме легочного локального амилоидоза позволяет предполагать, что оториноларингологи менее склонны учитывать такую, чаще терапевтическую проблему, как амилоидоз при проведении дифференциального диагноза в отличие от пульмонолога. Недостаточная информированность о проблеме, по-видимому, характерна и для врача-гистолога широкой практики. Об этом свидетельствуют длительные сроки установления диагноза амилоидоза после проведенной биопсии у значительной части больных. Следует учесть, что нарушение правил микротомирования и окрашивания препаратов амилоида красителем конго-красный в неопытных руках нередко дает неожиданные как ложноположительные, так и ложноотрицательные результаты, а в лабораториях часто отсутствует возможность проведения микроскопии в поляризованном свете, необходимой для диагностики амилоидоза. В среде врачей морфологов дискуссионным остается вопрос о необходимости включения в стандарт первичной обработки биопсийных препаратов метода окраски конго-красным. Показательно, что первичное предположение об опухолевом заболевании у больных с опухолевидным вариантом депозиции амилоида, нередко позволяющее рано провести биопсию пораженной ткани, не ускоряет диагностику амилоидоза по представленным выше соображениям, а также в силу настроенности морфолога на выявление клеточной атипии и недостаточное внимание к анализу межуточной ткани.
Объективные трудности диагностики демонстрирует локальный амилоидоз кожи, который у всех больных индуцировал инфильтративный диффузный процесс с развитием лихенификации кожи и тем самым имитировал более частые воспалительные заболевания кожи. По этой причине дерматологи часто склонны назначать лечение без проведения морфологического исследования.
Относительно быстрая диагностика ALL у больных с поражением конъюнктивы и тканей глаза, по-видимому, является результатом настойчивости больных, так как такие депозиты амилоида существенно влияют на функцию ведущего органа рецепции информации, а их локализация на лице становится причиной значительной социально-эстетической неудовлетворенности пациентов.
Опыт дифференциальной диагностики локального и системного AL-амилоидоза показывает необходимость широкого многопрофильного подхода к диагностике ALL с проведением обследования для подтверждения наличия плазмаклеточной дискразии с дифференцированием ее костномозгового и локального вариантов. С этой целью обязательным стандартом является приме нение высокочувствительных методов исследования крови и мочи с дополнением стандартного электрофореза методикой иммунофиксации сыворотки и мочи, количественного определения свободных легких цепей иммуноглобулинов в крови. Всегда следует стремиться также провести иммуногистохимическое определение клона плазматических клеток в ткани, что позволит обосновать применение современных методов лечения. В случае ALL и моноорганного поражения в дебюте системного AL полная уверенность в отсутствии или наличии системного варианта амилоидоза может быть достигнута только при дополнении иммунохимического исследования крови и мочи иммунофенотипированием костного мозга. Стандарт обследования должен включать также определение белка в моче, проведение эхокардиографии, осмотр невролога. Наблюдение за больными локальным ларинготрахеобронхиальным амилоидозом предполагает помимо регулярного эндоскопического обследования проведение также оценки показателей функции внешнего дыхания и структурных изменений в легких по данным компьютерной томографии с целью оценки скорости распространения амилоидоза и вовлечения легочной паренхимы.
В лечении больных ALL в первую очередь необходимо применять наименее травматичные методы иссечения тканей (радиоэксцизия, лазерная эксцизия и др.), после чего необходимо тщательное наблюдение пациента в течение не менее 4-5 лет для выявления рецидивов заболевания [23]. В случае рецидива ALL следует сделать вывод, что амилоидогенный клон плазмацитов в составе амилоида сохранил жизнеспособность. В этом случае эффективность хирургического лечения следует признать ограниченной, а дальнейшая терапия должна быть направлена на элиминацию патогенного клона плазматических клеток. В этом случае возможно применение химиотерапии, однако сравнение с плазмацитомой позволяет предполагать высокую радиочувствительность амилоидогенного клона плазмацитов, что с учетом локального характера амилоидоза создает большие перспективы для применения лучевой терапии у больных с рецидивом ALL. Проведение лучевой терапии, по-видимому является единственным способом лечения амилоидомы мочевого пузыря и амилоидоза кожи. В литературе имеются описания эффективного применения лучевой терапии при ларинготрахеобронхиальном амилоидозе [24]. В настоящее время остается актуальной задача дальнейшего изучения эффективности лучевой терапии при ALL разных локализаций.
Заключение
ALL является редким заболеванием, встречающимся с частотой приблизительно 1-2 случая на 1 миллион населения. Среди клинических вариантов ALL преобладают ларинготрахеобронхиальный амилоидоз и амилоидоз с поражением конъюнктивы и мягких тканей глазницы. Основной преградой для своевременной диагностики ALL является низкая информированность клиницистов разных специальностей и врачей-морфологов об этом заболевании. Своевременная диагностика амилоидоза предполагает внедрение стандартов, учитывающих необходимость расширения показаний к ранней биопсии. ALL необходимо дифференцировать от системного AL, в особенности при длительном течении последнего с преимущественно моноорганным поражением. В пользу ALL убедительно свидетельствуют отсутствие протеинурии и моноклональной гаммапатии. При этом результаты иммунохимического исследования крови и мочи следует дополнять иммунофенотипированием костного мозга с панелью антигенов-маркеров плазматических клеток и их клональности. Косвенным указанием на ALLамилоидоз может быть также нормальная толщина стенок миокарда и отсутствие признаков поражения автономной и периферической нервной системы. В лечении ALL в большинстве случаев достаточно применения методов хирургического удаления амилоида с применением малотравматичных методов иссечения тканей. Однако ALL рецидивирует у каждого четвертого больного, что требует поиска иных методов лечения. В частности, очевидное родство ALамилоидомы и плазмацитомы и известная чувствительность плазмацитомы к лучевой терапии оправдывают попытки применения у таких пациентов лучевой терапии с целью уничтожения локального амилоидогенного клона плазматических клеток.
Используемые источники
- Sipe JD, Benson MD, Buxbaum JN et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016;23(4):209–13.
- Reimann HA, Koucky RF, Eklund CM. Primary amyloidosis limited to tissue of mesodermal origin. Am J Pathol 1935;11(6):977–88.
- Mahmood S, Bridoux F, Venner CP et al. Natural history and outcomes in localised immunoglobulin light-chain amyloidosis: a long-term observational study. Lancet Haematol 2015;2(6):e241–50.
- Kourelis TV, Kyle RA, Dingli D et al. Presentation and outcomes of localized immunoglobulin light chain amyloidosis: The Mayo Clinic Experience. Mayo Clin Proc 2017;92(6):908–17.
- Carmona-Iragui M, Videla L, Lle A and Fortea J. Down syndrome, Alzheimer disease, and cerebral amyloid angiopathy: The complex triangle of brain amyloidosis. Dev Neurobiol 2019;Aug. 2019: dneu.22709.
- Yamada M, Naiki H. Cerebral amyloid angiopathy. Mol Biol Neurodeg Dis 2012;107:41-78.
- Серов В.В. Старческий амилоидоз: от тетрады Шварца до наших дней. РМЖ 1997;20:8. [Serov VV. Senile amyloidosis: from Schwarz tetrade to our days. RMJ 1997;20:8 (In Russ.)].
- Nagase T, Iwaya K, Zako T et al. Clinical and MRI characteristics and follow-up studies of insulin-derived amyloidosis. Amyloid 2019;26(suppl 1):156–57.
- Nilsson MR. Insulin amyloid at injection sites of patients with diabetes. Amyloid 2016;23(3):139–47.
- Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995;32(1):45–59.
- Рамеев В.В., Козловская Л.В., Рамеева А.С., и соавт. Особенности эволюции и прогностическое значение поражения сердца у больных системным AL-амилоидозом. Клин фармакол тер 2019;28(2):49–56. [Rameev VV, Kozlovskaya LV, Rameeva AS et al. Evolution and prognostic significance of heart involvement in patients with systemic AL-amyloidosis. Clin Pharmacol Ther = Klinicheskaya farmakologiya i terapiya 2019;28(2):49-56 (In Russ.)].
- Iida T, Yamashita K, Nakase H. Localized gastrointestinal AL amyloidosis. Clin Gastroenterol Hepatol 2018;16(9):e93.
- Kyle RA, Linos A, Beard CM et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992;79(7):1817–22.
- Pinney JH, Smith CJ, Taube JB et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol 2013;161:525–32.
- Multiple Myeloma: diagnosis and treatment. Gertz MA, Rajkumar SV (eds.). New York, NY: Springer New York. 2014:195–210.
- Liebross RH, Ha CS, Cox JD et al. Clinical course of solitary extramedullary plasmacytoma. Radiother Oncol 1999;52(3):245–9.
- Kyle RA, Therneau TM, Rajkumar SV et al. Incidence of multiple myeloma in Olmsted County, Minnesota: Trend over 6 decades. Cancer 2004;101(11): 2667–74.
- Dores GM, Landgren O, McGlynn KA et al. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol 2009;144(1):86–94.
- Rajkumar SV, Dimopoulos MA, Palumbo A et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014;15(12):e538–48.
- Alexiou C, Kau RJ, Dietzfelbinger H et al. Extramedullary plasmacytoma. Cancer 1999;85(11):2305–14.
- Thumallapally N, Meshref A, Mousa M, Terjanian T. Solitary plasmacytoma: Population-based analysis of survival trends and effect of various treatment modalities in the USA. BMC Cancer 2017;17(1):1–11.
- Bachar G, Goldstein D, Brown D et al. Solitary extramedullary plasmacytoma of the head and neck-Long-term outcome analysis of 68 cases. Head Neck 2008;30(8):1012–9.
- Piazza C, Cavaliere S, Foccoli P. et al. Endoscopic management of laryngo-tracheobronchial amyloidosis: a series of 32 patients. Eur Arch Oto-Rhino-Laryngol 2003;260(7):349–54.
- Truong MT, Kachnic LA, Grillone GA et al. Long-term results of conformal radiotherapy for progressive airway amyloidosis. Int J Radiat Oncol Biol Phys 2012;83(2):734–9.