АНЦА-ассоциированные интерстициальные заболевания легких: актуальные вопросы диагностики и лечения
В течение последних лет появляются данные о связи интерстициальных заболеваний легких (ИЗЛ) и АНЦА-ассоциированных васкулитов (ААВ), особенно у пациентов с микроскопическим полиангиитом с наличием антител к миелопероксидазе (МПО-АНЦА). В части случаев ИЗЛ является единственным или ведущим проявлением заболевания, определяющим объем терапии и прогноз жизни больного с ААВ. В статье обсуждаются современные концепции патогенеза, подходы к диагностике и лечению АНЦА-ассоциированных ИЗЛ, а также дальнейшие направления изучения данной аутоиммунной патологии.
Интерстициальные заболевания легких (ИЗЛ) характеризуются развитием про грессирующего диффузного воспалительного и/или фиброзирующего поражения легких и сходными клиникорентгенологическими проявлениями и гистологической картиной. В соответствии с классификацией, принятой Американским торакальным и Европейским респираторным обществами в 2013 г., к ИЗЛ относят более 200 заболеваний, как с установленной этиологией, так и идиопатических [1].
Антинейтрофильные цитоплазматические антитела (АНЦА) являются серологическим маркером АНЦА-ассоциированных васкулитов (ААВ), в том числе микроскопического полиангиита (МПА), гранулематоза с полиангиитом (ГПА) и эозинофильного гранулематоза с полиангиитом (ЭГПА) [2]. Поражение легких – одно из частых проявлений ААВ [3]: у 85-90% пациентов ГПА [4-6] и 25-55% с МПА [7-9] имеются отклонения при компьютерной томографии органов грудной клетки (КТ). При ЭГПА клинически значимые изменения в легких (за исключением бронхиальной астмы) встречаются реже [10]. Наиболее клинически значимыми вариантами поражения легких при ААВ, ассоциированными с неблагоприятным исходом, можно считать диффузное альвеолярное кровотечение (ДАК) и узловые образования различных размеров, в том числе с зонами распада [11].
В течение нескольких лет появляются данные о связи ИЗЛ и ААВ, особенно ассоциированным с антителами к миелопероксидазе (МПО-АНЦА) [11]. В части случаев при ААВ наблюдается изолированное поражение легких; кроме того, у небольшой части пациентов с идиопатическими интерстициальными пневмониями наличие АНЦА (чаще МПО-АНЦА, чем антитела к протеиназе-3 (ПР3-АНЦА)) не сопровождается симптомами системного васкулита или предшествует его развитию [12].
В настоящее время имеется ограниченное количество информации относительно подходов к диагностике и лечению АНЦА-ассоциированных интерстициальных заболеваний легких (АНЦА-ИЗЛ), что объясняется их относительной редкостью [12]. В то же время, наличие интерстициального поражения легких при ААВ может негативно влиять на прогноз заболевания и повышать риск летального исхода, что подчеркивает необходимость поиска его оптимального лечения. В статье представлен современный взгляд на классификацию АНЦА-ИЗЛ, а также обсуждаются вопросы патогенеза, диагностики, лечения и направления дальнейшего изучения данной патологии.
Эпидемиология и классификация
ИЗЛ встречаются чаще у пациентов с МПА (до 45%) и реже у пациентов с ГПА (менее 5%); при ЭГПА описаны лишь единичные случаи развития интерстициального поражения легких [13,14]. При АНЦА-ИЗЛ отмечается значительное преобладание МПО-АНЦА (46-71%) по сравнению с ПР3АНЦА (0-29%) [13,15-20].
Распространенность ИЗЛ при ААВ зависит от географического региона: в Европе она составляет 2-3%, что существенно ниже по сравнению со странами Азии (2839%) [21,22]. Это может быть отчасти связано с большей частотой выявления МПО-АНЦА в азиатской популяции [11].
Возрастная структура АНЦА-ИЗЛ несколько отличается от таковой в общей группе ААВ. АНЦА-ИЗЛ, как правило, развиваются у пациентов старше 65 лет, хотя единичные случаи описаны даже у детей [11]. В ряде исследований средний возраст на момент диагностики МПА-ассоциированного ИЗЛ (МПА-ИЗЛ) превышал таковой для общей когорты пациентов с МПА (66 и 55 лет, соответственно) и был сопоставим с возрастом пациентов в дебюте идиопатического легочного фиброза (ИЛФ) [7,8]. В нескольких сериях клинических случаев МПА-ИЗЛ наблюдалось незначительное преобладание мужчин (60-65%), однако это отмечалось не во всех наблюдениях [15,23]. Многофакторный анализ в одном из исследований, включавших 62 пациента с ААВ-ИЗЛ, показал, что мужской пол и возраст старше 65 лет были независимыми предикторами развития легочного поражения [24].
Среди АНЦА-ИЗЛ можно выделить три основные группы. Первая включает пациентов с развернутой клинической картиной МПА, развившейся до поражения легких (8-21%); во вторую входят пациенты с ИЗЛ, дебютировавшим одновременно с системным васкулитом (36-67%); третья объединяет АНЦА-позитивных пациентов с ИЗЛ без признаков поражения других органов (14-85%) [11,17,25]. Последний клинический сценарий представляет особый диагностический интерес в связи с возможностью развития в последующем ААВ в срок от нескольких месяцев до 12 лет [11]. В одном исследовании на момент установления диагноза ИЛФ у 4,0% из 504 пациентов определялись МПОАНЦА, у 3,2% – ПР3-АНЦА. В течение последующих 5 лет сероконверсия произошла еще у 11% исходно АНЦА-негативных пациентов, а у одной четверти больных с МПО-АНЦА развился МПА [25]. Наличие других типов аутоантител, в частности ревматоидного фактора (РФ), повышение СОЭ >40 мм/ч, эозинофилия лаважной жидкости и большой объем поражения легочной ткани по данным КТ служили прогностическими факторами АНЦА-сероконверсии [26].
Патогенез
К настоящему времени предложены несколько потенциальных механизмов развития АНЦА-ИЗЛ. Согласно одной из гипотез, формирование интерстициального легочного фиброза при ААВ может быть следствием рецидивирующих эпизодов ДАК [27]. В пользу данной концепции свидетельствует наличие гистологических признаков острого и/или хронического кровотечения более чем у половины пациентов с АНЦА-ИЗЛ [28]. Важно отметить, что у большинства из них отсутствуют анамнестические данные о ДАК, что указывает на вероятное развитие субклинических эпизодов кровотечений [28,29]. Еще одним подтверждением данной гипотезы являются обнаружение в ткани легких значительного количества сидерофагов – гистологических маркеров хронического альвеолярного кровотечения – у пациентов с АНЦА-ИЗЛ и отсутствие данного признака у пациентов с ИЗЛ в рамках других системных аутоиммунных заболеваний, таких как воспалительные миопатии и системная склеродермия [30].
Согласно второй гипотезе, МПО-АНЦА (но не ПР3АНЦА) могут сами по себе играть роль в патогенезе ИЗЛ [31]. В одном исследовании было показано, что активация нейтрофилов антителами к МПО приводила к выработке большого количества окислительных соединений. К ним, в частности, относится гипохлорит-анион, способный стимулировать пролиферацию фибробластов in vitro. Кроме того, МПО-АНЦА могут способствовать повреждению легочной ткани путем локального высвобождения протеолитических ферментов активированными нейтрофилами. Некоторые из этих ферментов, в частности эластаза, могут вызывать развитие легочного фиброза в опытах на животных [32].
Повреждение, вызванное внеклеточными нейтрофильными ловушками, выделяющимися АНЦА-активированными нейтрофилами при их гибели путем нетоза, также может также вносить вклад в легочный патологический процесс. Внеклеточные нейтрофильные ловушки обладают способностью активировать легочные фибробласты и стимулировать их дифференцировку в миофибробласты – один из основных типов клеток, активно вовлеченных в развитие интерстициального легочного фиброза [33].
Еще одна концепция патогенеза АНЦА-ИЗЛ согласуется с фактом развития ИЗЛ до дебюта ААВ и основывается на том, что наличие ИЗЛ само по себе предрасполагает к появления МПО-АНЦА [24]. У пациентов с ИЛФ в лаважной жидкости в большинстве случаев увеличено количество нейтрофилов [34]. При активации нейтрофилы начинают экспрессировать МПО на своей плазматической мембране, что при наличии тканевого воспаления может запускать развитие аутоиммунной реакции и приводить к секреции МПО-АНЦА, а затем и развитию ААВ. В пользу данной гипотезы свидетельствуют появление МПО-АНЦА и развитие МПА у части пациентов с ИЛФ. В этой связи представляет интерес тот факт, что при ААВ, как и при ИЛФ, одним из факторов риска развития ИЗЛ является курение (частота его составила 59% и 23% у пациентов с МПА, у которых отмечалось и отсутствовало ИЗЛ, соответственно) [25].
В то же время, несмотря на ряд сходств, некоторые механизмы развития ИЛФ не играют значимой роли в патогенезе поражения легких при ААВ. В частности, ключевая роль апоптоза альвеолярных эпителиоцитов вследствие длительного воздействия повреждающих факторов, а также эпителиально-мезенхимального перехода не была подтверждена при АНЦА-ассоциированном интерстициальном легочном фиброзе. И наоборот, роль нейтрофилов и системы комплемента в развитии ИЛФ представляется незначительной по сравнению с таковой при АНЦА-ИЗЛ [35].
Клиническаякартина
Основными симптомами у пациентов с АНЦА-ИЗЛ является одышка (50-73%) и малопродуктивный кашель (21-60%) [36,37]. Кровохарканье (5%) и конституциональные проявления, такие как лихорадка (31%) и снижение массы тела (5%), наблюдаются реже [36]. В ранее опубликованных исследованиях не было выявлено достоверной корреляции между титром АНЦА и тяжестью ИЗЛ [36]. В ряде наблюдений клинические проявления у пациентов с фиброзирующими ИЗЛ и наличием АНЦА не отличались от таковых при ИЛФ [36,37].
В то же время, у пациентов с МПА-ИЗЛ в дебюте обычно преобладают конституциональные симптомы (около 80% случаев) – недомогание (31-63%), лихорадка (52-90%) и снижение массы тела (52-58%), а также внелегочные проявления основного заболевания (70100%) [38,39]. Легочные симптомы включают в себя прогрессирующую одышку (30-100%), кровохарканье (21-49%) и кашель (23-84%) [29,38,39]. Следует отметить, что у пациентов с МПА-ИЗЛ реже выявляют признаки системного воспаления, что выражается в более низком уровне СОЭ, более высоком уровне гемоглобина, и, что важно, в меньшей частоте таких проявлений васкулита, как ДАК, поражение периферической нервной системы и почек [29,40].
Диагностика
Повышение уровня СОЭ и содержания С-реактивного белка (СРБ) в дебюте заболевания у пациентов с МПАИЗЛ отмечается в 95% и 73-79% случаев, соответственно [17]. Более чем у 60% из них выявляют также изменения в общем анализе мочи [25]. В то же время, значительное повышение уровня воспалительных маркеров редко встречается при изолированном АНЦАИЗЛ. При сравнении данной группы с ИЛФ в большинстве исследований различий по уровню СРБ не обнаружено [36,37]. При исследовании сывороточных маркеров легочного повреждения у пациентов с ААВ повышение уровня Krebs von der Lungen (KL)-6 ассоциировалось с наличием ИЗЛ [41]. При ААВ-ИЗЛ отмечается четкая корреляция между наличием МПО-АНЦА и ИЗЛ; у пациентов с ПР3-АНЦА развитие ИЗЛ наблюдается значительно реже [24]. Кроме того при наличии ПР3-АНЦА у пациентов с ИЛФ ни в одном случае не было отмечено развития ААВ [25].
При АНЦА-ИЗЛ чаще всего развиваются вентиляционные нарушения рестриктивного типа – снижение форсированной жизненной емкости легких (ФЖЕЛ) и диффузионной способности легких (DLCO), хотя примерно в трети случаев наблюдается умеренная бронхообструкция [15,36,38]. В динамике отмечается тенденция к дальнейшему снижению вентиляционных параметров по мере прогрессирования ИЗЛ. В одном исследовании у больных с ААВ-ИЗЛ в течение 5 лет отмечалось снижение жизненной емкости легких и (ЖЕЛ) и DLCO на 23% и 46%, соответственно, по сравнению с исходными значениями [42].
При исследовании бронхоальвеолярной лаважной жидкости (БАЛЖ) у пациентов с АНЦА-ИЗЛ выявляют нейтрофилию (40-87%), реже – лимфоцитоз и эозинофилию (20% и 26%, соответственно) [15,36]. Признаки острого или хронического альвеолярного кровотечения обнаруживают в половине случаев [15]. По данным ряда авторов, при АНЦА-ИЗЛ количество нейтрофилов и эозинофилов в БАЛЖ было выше, чем при ИЛФ, однако эти результаты подтверждаются не всеми исследованиями [15,37,43].
Основными изменениями на КТ, ассоциированными с ААВ, являются уплотнение по типу "матового стекла" (23-100%), ретикулярные изменения (41-77%), утолщение междолькового интерстиция (41-71%), зоны консолидации (0-78%) и "сотового легкого" (23-100%). Реже выявляют узловые образования в паренхиме (0-45%) и кисты (27%) [22,39,42]. Может наблюдаться поражение дыхательных путей в форме бронхиолита (55%), утолщения стенок бронхов (44%) и бронхоэктазов (32100%). У большинства пациентов (50-100%) изменения в легких носят симметричный характер [38,42,44].
Согласно международным классификационным критериям идиопатических интерстициальных пневмоний, наиболее частым КТ-паттерном ААВ-ИЗЛ, как при наличии МПО-АНЦА, так и ПР3-АНЦА, является обычная интерстициальная пневмония (ОИП; 38-63%), характеризующаяся ретикулярными изменениями, преимущественно в задних и нижних отделах легких, в сочетании с формированием зон "сотового легкого" и уменьшением объема нижних долей. Несколько реже отмечается неспецифическая интерстициальная пневмония (НСИП; 7-31%) и десквамативная интерстициальная пневмония (14%) (рис. 1). У части пациентов (преимущественно курящих мужчин) описано развитие интерстициального фиброза в сочетании с эмфиземой легких (до 21%) [6,38,39].
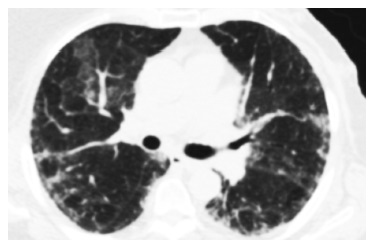
Следует отметить, что в 4-40% случаев изменения в легких при АНЦА-ИЗЛ не соответствуют какому-либо конкретному паттерну интерстициальной пневмонии [36]. В спорных случаях для определения показаний к иммуносупрессивной терапии и ее объема требуется гистологическое подтверждения диагноза. В связи с невозможностью проведения видео-ассоциированной торакоскопической биопсии легкого у части пациентов из-за выраженных вентиляционных нарушений и дыхательной недостаточности может быть также рассмотрено выполнение криобиопсии легкого [24].
Как и в случае рентгенологической картины, основным гистологическим паттерном АНЦА-ИЗЛ является ОИП (46-100%), на втором месте по частоте находится НСИП (7-31%) [36,37]. Следует отметить, что несмотря на невысокую распространенность НСИП, в части случаев при основном паттерне ОИП у пациентов также выявляли отдельные зоны с НСИП-подобной гистологической картиной. Кроме того, в отличие от ОИПИЛФ, при АНЦА-ОИП чаще наблюдаются признаки интерстициального воспаления, поражения мелких дыхательных путей, а также лимфоидные фолликулы [36]. Интересно, что признаки активного васкулита редко определяются в биоптатах легкого у пациентов с АНЦА-ИЗЛ [37].
ИЗЛ при ААВ следует дифференцировать с ДАК – одним из наиболее опасных проявлений системного васкулита [11]. Для КТ-картины ДАК в острой стадии характерно наличие диффузно расположенных зон матового стекла, которые в ряде случаев могут занимать большую часть объема легочной паренхимы, с визуализирующимися заполненными сегментарными и субсегментарными бронхами. Скопления крови в полости альвеол могут также формировать различные по размеру зоны консолидации. В подостром периоде (через 2-3 дня после эпизода кровотечения) для ДАК характерно утолщение междолькового и внутридолькового интерстиция, в ряде случаев – на фоне сохраняющегося уплотнения по типу матового стекла (КТ-симптом "булыжной мостовой"). В исходе ДАК в легких могут формироваться множественные нечеткие центрилобулярные очаги, соответствующие интраальвеолярным скоплениям сидерофагов; вследствие обильных рецидивирующих ДАК может развиться грубый интерстициальный легочный фиброз [6,11].
С учетом активной комбинированной иммуносупрессивной терапии, проводимой большинству пациентов с ААВ, в круг дифференциального диагноза следует включать инфекционные осложнения, в первую очередь, обусловленные оппортунистическими микроорганизмами. Так, пневмония, вызванная Pneumocystis jiroveci, характеризуется появлением обширных зон матового стекла в легочной ткани и может имитировать обострение ИЗЛ (рис. 2) [45].
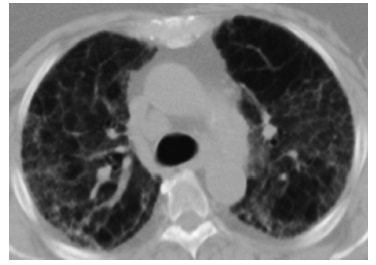
Лекарственное поражение легких, особенно при длительном приеме и высокой кумулятивной дозе цитостатических препаратов, также должно рассматриваться в качестве причины развития ИЗЛ у пациентов с ААВ [11].
Лечение и прогноз
Согласно современным клиническим рекомендациям, лечение ААВ включает индукцию ремиссии с использованием высокоактивных иммуносупрессивных препаратов (высоких доз ГКС в сочетании с циклофосфамидом или ритуксимабом) с последующим переходом на прием поддерживающей терапии для предотвращения рецидива [46]. В то же время, стандартные протоколы терапии не учитывают особенности клинической картины заболевания в отдельных подгруппах пациентов, в том числе при АНЦА-ИЗЛ. Использование иммуносупрессивной терапии при интерстициальном поражении легких в рамках ААВ отчасти основывается на ее доказанной эффективности при ААВ и ИЗЛ, ассоциированных с рядом системных заболеваний соединительной ткани, в частности системной склеродермией и воспалительными миопатиями [11].
Тем не менее, полученные к настоящему моменту данные об эффективности иммуносупрессивной терапии при лечении ААВ-ИЗЛ носят противоречивый характер. С одной стороны, в ряде работ была выявлена клиническая или рентгенологическая положительная динамика у 80% пациентов с ААВ-ИЗЛ, получавших иммуносупрессивную терапию [36]. В исследовании С. Comarmond и соавт. 3-летняя выживаемость пациентов, получавших комбинированную терапию ГКС в сочетании с циклофосфамидом, составила 94% и превышала таковую у больных, которым проводилась монотерапия глюкокортикостероидами – 65% [17]. Имеются указания на зависимость эффективности иммуносупрессивной терапии от паттерна легочного поражения: у пациентов с КТ-картиной НСИП чаще отмечается положительная динамика легочного поражения в ответ на иммуносупрессивную терапию, тогда как у пациентов с ОИП чаще наблюдается прогрессирование ИЗЛ, несмотря на проводимое лечение, что позволяет провести аналогию с ИЛФ [11].
Нерешенным остается вопрос об эффективности иммуносупрессивной терапии при изолированных АНЦА-ИЗЛ, так как большая часть информации была получена из ретроспективных исследований в неоднородных выборках [12]. В ряде исследований было показано, что прогноз при АНЦА-ИЗЛ достоверно не отличается от такового при ИЛФ (средняя выживаемость – 2,5 и 3,5 года с момента установления диагноза, соответственно), вне зависимости от объема проводимой иммуносупрессивной терапии [47]. И наоборот, другие работы продемонстрировали, что выживаемость пациентов с АНЦА-ИЗЛ занимает промежуточное положение между ИЛФ и ААВ без поражения легких [40]. На основании имеющихся данных в 2019 г. группой исследователей из клиники Мэйо (Рочестер, США) была предложена эмпирическая схема лечения МПО-АНЦА-ассоциированного ИЗЛ (табл. 1) [12].
| КТ-паттерн поражения легких |
Клинико-лабораторная картина | Лечение |
|---|---|---|
| Примечание: ГКС – глюкокортикостероиды, ММФ – микофенолата мофетил, АЗА – азатиоприн | ||
| ОИП | Изолированное наличие МПО-АНЦА | Наблюдение + ежемесячное проведение общего анализа мочи для исключения гематурии |
| МПО-АНЦА + повышение уровня воспалительных маркеров | Наблюдение + ежемесячное проведение общего анализа мочи для исключения гематури | |
| МПО-АНЦА и МПА | Стандартная терапия МПА | |
| НСИП | Изолированное наличие МПО-АНЦА | ГКС + ММФ/АЗА |
| МПО-АНЦА + повышение уровня воспалительных маркеров | ГКС + ММФ/АЗА | |
| МПО-АНЦА и МПА | Стандартная терапия МПА | |
В будущем для лечения АНЦА-ИЗЛ могут быть использованы антифибротические лекарственные препараты, такие как пирфенидон и нинтеданиб, замедляющие прогрессирование и улучшающие выживаемость пациентов с ИЛФ. Данное предположение основано на том, что в 2019 г. завершено клиническое исследование, продемонстрировавшее эффективность антифибротической терапии у пациентов с фиброзирующими интерстициальными заболеваниями легких, не соответствующими ИЛФ, включая поражение легких в рамках системных заболеваний соединительной ткани [48]. В настоящее время Французской группой по изучению васкулитов проводится открытое исследование эффективности антифибротической терапии у пациентов с ААВ-ИЗЛ (NCT03385668) [24].
Поражение легких может вносить негативный вклад в долгосрочный прогноз пациентов с МПА. Так, в рядеисследований было выявлено повышение смертности в 2-4 раза в подгруппе пациентов ААВ-ИЗЛ по сравнению таковой у больных ААВ без поражения легких [24]. Средняя продолжительность жизни после установления диагноза ААВ-ИЗЛ составляет 3,5-6 лет при уровне 5-летней выживаемости 29-60%, что лишь незначительно превышает соответствующие значения при ИЛФ [36,37]. Основными причинами смерти при ААВ-ИЗЛ являются инфекционные осложнения, обострение ИЗЛ, а также прогрессирующее поражение легких с развитием терминальной дыхательной недостаточности [11].
В то же время в других исследованиях не было выявлено достоверных отличий между выживаемостью пациентов с ААВ и ААВ-ИЗЛ. Одной из вероятных причин этого может быть относительно короткий период наблюдения [24]. Вместе с тем, в когорте из 504 пациентов с ИЛФ 5- и 10-летняя смертность у АНЦАпозитивных пациентов была достоверно выше (61,3% и 85,7%), чем у АНЦА-негативных (37,6% и 70,5%). В данном исследовании, наличие ПР3-АНЦА (по сравнению с МПО-АНЦА), возраст более 65 лет, а также исходное значение DLCO менее 70% были ассоциированы с повышенной смертностью [25].
Заключение
ИЗЛ является одним из вариантов поражения легких при ААВ, оказывающим негативное влияние на течение и прогноз заболевания. У части пациентов ИЗЛ является первым проявлением болезни, развивающимся до появления других симптомов системного васкулита. Таким образом, исследование уровня АНЦА должно проводиться всем пациентам с идиопатическими интерстициальными пневмониями в рамках дифференциального диагноза.
Обсуждая подходы к терапии и тактику дальнейшего наблюдения, АНЦА-ИЗЛ можно разделить на две основные группы. К первой относятся интерстициальные поражения легких при наличии развернутой картины системного васкулита (как правило, МПА), требующего проведения стандартной иммуносупрессивной терапии и наблюдения ревматологами. Вторую составляют случаи ИИП, установленной в качестве первичного диагноза и сочетающейся с АНЦА. У таких пациентов важно оценить уровень воспалительных маркеров, а также провести активный поиск внелегочных, в том числе субклинических, проявлений ААВ, в частности, поражения почек, нервной системы, кожи, дыхательных путей, суставов. При наличии классификационных критериев ААВ необходимо проводить стандартную иммуносупрессивную терапию. Если данные в пользу ААВ отсутствуют, то назначение иммуносупрессивной терапии зависит от КТ-паттерна поражения легких. В части случаев обсуждается использование антифибротических препаратов.
Учитывая относительную редкость патологии, для получения достоверных данных об ее распространен ности в различных регионах, эффективности иммуносупрессивной и антифибротической терапии, определения оптимальной тактики ведения больных с АНЦА-ИЗЛ, требуется создание международных регистров пациентов и проведение проспективных рандомизированных клинических исследований. Кроме того, для сопоставления АНЦА-ИЗЛ с другими вариантами аутоиммунных поражений легких представляется обоснованным включение АНЦА в серологические критерии интерстициальной пневмонии с аутоиммунными признаками [49].
Используемые источники
- Travis WD, Costabel U, Hansell DM, et al. An Official American Thoracic Society/European Respiratory Society Statement: Update of the International Multidisciplinary Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2013:188(6):733–48.
- Новиков П.И., Зыкова А.С., Смитиенко И.О., Моисеев С.В. Лечение АНЦА-ассоциированных васкулитов: рекомендации EULAR/ERA-EDTA 2016 года. Клин фармакол тер 2017;1:80-7 [Novikov PI, Zykova AS, Smitienko IO, Moiseev S. Treatment for ANCA-associated vasculitides: 2016 EULAR/ERAEDTA guidelines. Klinicheskaya farmakologiya i terapiya = Clinical Pharma co logy and Therapy 2016;26(1):80-7 (In Russ.)].
- Nachman PH, Henderson AG. Pathogenesis of lung vasculitis. Semin Respir Crit Care Med 2011;32:245–53.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992;116:488–98.
- Anderson G, Coles ET, Crane M, et al. Wegener’s granuloma. A series of 265 British cases seen between 1975 and 1985. A report by a sub-committee of the British Thoracic Society Research Committee. Q J Med 1992;83:427–38.
- Comarmond C, Cacoub P. Granulomatosis with polyangiitis (Wegener): clinical aspects and treatment. Autoimmun Rev 2014;13:1121–5.
- Schirmer JH, Wright MN, Vonthein R, et al. Clinical presentation and long-term outcome of 144 patients with microscopic polyangiitis in a monocentric German cohort. Rheumatology (Oxford) 2016;55:71–9.
- Guillevin L, Durand-Gasselin B, Cevallos R, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42: 421–30.
- Villiger PM, Guillevin L. Microscopic polyangiitis: clinical presentation. Auto immun Rev 2010;9:812–9.
- Cottin V, Bel E, Bottero P, Dalhoff K, et al. Respiratory manifestations of Eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Eur Respir J 2016;48(5):1429-41.
- Alba MA, Flores-Suárez LF, Henderson AG. Interstital lung disease in ANCA vasculitis. Autoimmun Rev 2017;16(7):722–9.
- Thompson GE, Specks U. Update on the management of respiratory manifestations of the antineutrophil cytoplasmic antibodies-associated vasculitides. Clin Chest Med 2019;40(3):573-82.
- Chen M, Yu F, Zhang Y, et al Antineutrophil cytoplasmic autoantibody-associated vasculitis in older patients. Medicine (Baltimore) 2008;87:203–9.
- Новиков П.И., Моисеев С.В., Буланов Н.М., Макаров Е.А. Современные подходы к терапии АНЦА-ассоциированных системных васкулитов. Клиническая нефрология 2014;1:42-9. [Novikov PI, Moiseev SV, Bulanov NM, Makarov EA. Current treatment of ANCA-associated systemic vasculitides. Klinicheskaya nefrologiya = Clinical Nephrology 2014;1,42-9 (In Russ.)].
- Foulon G, Delaval P, Valeyre D, et al. ANCA-associated lung fibrosis: analysis of 17 patients. Respir Med 2008;102:1392–8.
- Tanaka T, Otani K, Egashira R, et al. Interstitial pneumonia associated with MPO-ANCA: clinicopathological features of nine patients. Respir Med 2012;106:1765–70.
- Comarmond C, Crestani B, Tazi A, et al. Pulmonary fibrosis in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: a series of 49 patients and review of the literature. Medicine (Baltimore) 2014;93:340–9.
- Katsumata Y, Kawaguchi Y, Yamanaka H. Interstitial lung disease with ANCAassociated vasculitis. Clin Med Insights Circ Respir Pulm Med 2015;9:51–6.
- Hirayama K, Kobayashi M, Usui J, et al. Pulmonary involvements of anti-neutrophil cytoplasmic autoantibody-associated renal vasculitis in Japan. Nephrol Dial Transplant 2015;30(Suppl. 1):i83–93.
- Hozumi H, Enomoto N, Oyama Y, et al. Clinical implication of proteinase-3antineutrophil cytoplasmic antibody in patients with idiopathic interstitial pneumonias. Lung 2016;194:235–42.
- Sada K, Yamamura M, Harigai M, et al. Classification and characteristics of Japanese patients with antineutrophil cytoplasmic antibody-associated vasculitis in a nationwide, prospective, inception cohort study. Arthritis Res Ther 2014;16(2):R101.
- Yamagata M, Ikeda K, Tsushima K, et al. Prevalence and responsiveness to treatment of lung abnormalities on chest computed tomography in patients with microscopic polyangiitis: a multicenter, longitudinal, retrospective study of one hundred fifty consecutive hospital-based Japanese patients. Arthritis Rheumatol 2016;68:713–23.
- Nada AK, Torres VE, Ryu JH, et al. Pulmonary fibrosis as an unusual clinical manifestation of a pulmonary-renal vasculitis in elderly patients. Mayo Clin Proc 1990;65:847–56.
- Maillet T, Goletto T, Beltramo G, et al. Usual interstitial pneumonia in ANCAassociated vasculitis: A poor prognostic factor. J Autoimmun 2019 Sep 27:102338.
- Kagiyama N, Takayanagi N, Kanauchi T, et al. Antineutrophil cytoplasmic antibody-positive conversion and microscopic polyangiitis development in patients with idiopathic pulmonary fibrosis. BMJ Open Resp Res 2015;2: e000058.
- Ando M, Miyazaki E, Ishii T, et al. Incidence of myeloperoxidase anti-neutrophil cytoplasmic antibody positivity and microscopic polyangitis in the course of idiopathic pulmonary fibrosis. Respir Med 2013;107: 608–15.
- Birnbaum J, Danoff S, Askin FB, et al. Microscopic polyangiitis presenting as a “pulmonary-muscle” syndrome: is subclinical alveolar hemorrhage the mechanism of pulmonary fibrosis? Arthritis Rheum 2007;56:2065–71.
- Travis WD, Hoffman GS, Leavitt RY, et al. Surgical pathology of the lung in Wegener’s granulomatosis. Review of 87 open lung biopsies from 67 patients. Am J Surg Pathol 1991;15:315–33.
- Fernandez Casares M, Gonzalez A, Fielli M, et al. Microscopic polyangiitis associated with pulmonary fibrosis. Clin Rheumatol 2015;34:1273–7.
- Schnabel A, Reuter M, Csernok E, et al. Subclinical alveolar bleeding in pulmonary vasculitides: correlation with indices of disease activity. Eur Respir J 1999;14:118–24.
- Guilpain P, Chereau C, Goulvestre C, et al. The oxidation induced by antimyeloperoxidase antibodies triggers fibrosis in microscopic polyangiitis. Eur Respir J 2011;37:1503–13.
- Foucher P, Heeringa P, Petersen AH, et al. Antimyeloperoxidase-associated lung disease. An experimental model. Am J Respir Crit Care Med 1999;160:987–94.
- Yoshida M, Yamada M, Sudo Y, et al. Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrology (Carlton) 2016;21:624–9.
- Borie R, Crestani B. Antineutrophil сytoplasmic antibody-associated lung fibrosis. Semin Respir Crit Care Med 2018;39:465-70.
- Gindre D, Peyrol S, Raccurt M, et al. Fibrosing vasculitis in Wegener’s granulomatosis: ultrastructural and immunohistochemical analysis of the vascular lesions. Virchows Arch 1995;427:385–93.
- Nozu T, Kondo M, Suzuki K, et al. A comparison of the clinical features of ANCA-positive and ANCA-negative idiopathic pulmonary fibrosis patients. Respiration 2009;77:407–15.
- Hosoda C, Baba T, Hagiwara E, et al. Clinical features of usual interstitial pneumonia with anti-neutrophil cytoplasmic antibody in comparison with idiopathic pulmonary fibrosis. Respirology 2016;21:920–6.
- Hervier B, Pagnoux C, Agard C, et al. Vasculitis study, pulmonary fibrosis associated with ANCA-positive vasculitides. Retrospective study of 12 cases and review of the literature. Ann Rheum Dis 2009;68:404–7.
- Tzelepis GE, Kokosi M, Tzioufas A, et al. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J 2010;36:116–21.
- Flores-Suarez LF, Ruiz N, Saldarriaga Rivera LM, et al. Reduced survival in microscopic polyangiitis patients with pulmonary fibrosis in a respiratory referral centre. Clin Rheumatol 2015;34:1653–4.
- Iwata Y, Wada T, Kitagawa K, et al. Serum levels of KL6 reflect disease activity of interstitial pneumonia associated with ANCA-related vasculitis. Intern Med 2001;40:1093-7.
- Arulkumaran N, Periselneris N, Gaskin G, et al. Interstitial lung disease and ANCA-associated vasculitis: a retrospective observational cohort study. Rheumatology (Oxford) 2011;50:2035–43.
- Ando Y, Okada F, Matsumoto S, et al. Thoracic manifestation of myeloperoxidase antineutrophil cytoplasmic antibody (MPO-ANCA)-related disease. CT findings in 51 patients. J Comput Assist Tomogr 2004;28:710–6.
- Huang H, Wang YX, Jiang CG, et al. A retrospective study of microscopic polyangiitis patients presenting with pulmonary fibrosis in China. BMC Pulm Med 2014;14:8.
- Wolfe RM, Peacock JE. Pneumocystis pneumonia and the rheumatologist: which patients are at risk and how can PCP be prevented? Curr Rheumatol Rep 2017;19(6): 35.
- Holle JU, Gross WL. Treatment of ANCA-associated vasculitides (AAV). Autoimmun Rev 2013;12:483–6.
- Homma S, Matsushita H, Nakata K. Pulmonary fibrosis in myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitides. Respirology 2004;9: 190–6.
- Flaherty KR, Wells AU, Cottin V. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019; 381(18):1718-27.
- Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. EurRespir J 2015;46:976–87.