Поражение верхних дыхательных путей при АНЦА-ассоциированных васкулитах
АНЦА-ассоциированные васкулиты (ААВ) относят к группе системных воспалительных заболеваний, характеризующихся некротизирующим поражением преимущественно мелких сосудов и частым выявлением антинейтрофильных цитоплазматических антител (АНЦА) в сыворотке крови. Поражение верхних дыхательных путей (ВДП) часто встречается при ААВ и обычно сочетается с другими проявлениями заболевания – поражением почек, легких и кожи. Однако у 25% больных гранулематозом с полиангиитом наблюдается изолированное воспаление ВДП. Варианты поражения ВДП при ААВ включают в себя язвенно-некротические изменения слизистых оболочек, полипы в полости носа, деструкцию костных и хрящевых структур и стеноз трахеи. В обзоре литературы обсуждаются различные проявления поражения ВДП при ААВ, современные методы их диагностики и лечения. Поражение ВДП зачастую развивается первым и долгое время может оставаться единственным проявлением системного заболевания, поэтому соответствующая настороженность необходима не только среди ревматологов, но и терапевтов, врачей общей практики и оториноларингологов.
АНЦА-ассоциированные васкулиты (ААВ) – это группа системных аутоиммунных заболеваний, характеризующихся воспалительным некротизирующим поражением стенок преимущественно мелких (капилляры и венулы) и, существенно реже, средних сосудов различных органов , а также частым наличием антинейтрофильных цитоплазматических аутоантител (АНЦА) в сыворотке крови. К первичным ААВ относят гранулематоз с полиангиитом (ГПА), микроскопический полиангиит (МПА) и эозинофильный гранулематоз с полиангиитом (ЭГПА), а также АНЦА-ассоциированный васкулит с изолированным поражением почек [1]. Выделяют также вторичные васкулиты, которые могут сопровождаться образованием АНЦА, например, лекарственный васкулит, вызванный приемом кокаина, смешанного с левамизолом [2]. Язвенно-некротическое поражение кожи возникает спустя несколько дней после употребления левамизола, при этом гистологически определяется лейкоцитокластический васкулит с фибриноидным некрозом сосудистых стенок и фибриновыми тромбами в мелких сосудах кожи. Поражение слизистой оболочки ВДП связано в первую очередь не с васкулитом, а с местным раздражающим действием кокаина в составе смеси.
Поражение верхних дыхательных путей встречается при всех ААВ, а у части пациентов может быть первым и в течение длительного времени единственным проявлением системного заболевания. Так, по данным J. Wojciechowska и соавт., от появления первых симптомов поражения ВДП до установления диагноза ГПА или МПА в среднем проходит 14,5 месяцев [3].
Варианты поражения ВДП при ААВ
Гранулематоз с полиангиитом. ГПА (ранее гранулематоз Вегенера) – это системный васкулит, характеризующийся наличием гранулематозного воспаления и развитием некротического поражения мелких и средних сосудов [4]. Заболевание может поражать все органы, однако наиболее частыми мишенями оказываются верхние и нижние дыхательные пути, а также почки [5]. ГПА обычно развивается у людей среднего и старшего возраста (40–50 лет), в равной степени у мужчин и женщин [6], хотя среди российских пациентов преобладали женщины (60%). Средний возраст больных на момент начала заболевания не превышает 40 лет [7].
Заболеваемость ГПА зависит от географического региона. Например, в Великобритании заболеваемость ГПА составляет 14 случаев на один миллион населения в год, а распространенность – 134,9 случаев на миллион населения [8]. В Новой Зеландии заболеваемость ГПА находится на том же уровне – 15 случаев на миллион населения [9]. В странах Азии заболеваемость ГПА в целом ниже и варьируется от 0,37 до 2,1 случаев на миллион населения в год, а распространенность заболевания в Китае составляет 19,4 случая на миллион [10]. К сожалению, имеющихся на данный момент российских данных по эпидемиологии ААВ недостаточно для объективной оценки заболеваемости и распространенности в нашей стране.
При ГПА поражение органов головы и шеи, включая ЛОР-органы, отмечается в 80-95% случаев и чаще всего является первым симптомом васкулита [11]. По данным исследования A. Fauci и соавт., за 21 год наблюдения поражение ВДП развилось у 95% пациентов с ГПА [12]. Вовлечение в патологический процесс носа и околоносовых пазух отмечается у 60-90% пациентов с ГПА, в странах Азии этот показатель составляет 79-91% [10]. В российской когорте пациентов с ГПА поражение ВДП наблюдалось более чем в 80% случаев [7]. Зачастую поражение ВДП указывает на высокую активность системного васкулита [13]. Кроме того, у 25% пациентов с ГПА развивается локальная форма заболевания, ограничивающаяся исключительно симптомами поражения носа и околоносовых пазух [14].
Основные проявления активного ГПА с поражением ВДП включают в себя наличие корок в носовых ходах (69%), симптомы хронического риносинусита (61%), затруднение носового дыхания (58%) и кровянистые выделения из носа (52%) [15,16], а также неприятно пахнущие выделения из носа, рецидивирующие носовые кровотечения, гипосмию и аносмию, слезотечение, вызванное обструкцией носослезных протоков. В ретроспективном 20-летнем исследовании C. MoralesAngulo и соавт. представлены несколько иные данные: поражение носа и придаточных пазух наблюдалось у 52% пациентов с ГПА и в основном было представлено хроническим риносинуситом (32%) [17]. Заложенность носа часто является первым симптомом поражения ВДП, а гипосмия или аносмия обычно развиваются уже при наличии обширного отека слизистой оболочки [18]. Слезотечение также нередко отмечается в начале заболевания и может быть вызвано как гранулематозным поражением носослезного протока и слезного мешка, так и присоединением инфекции [19,20].
Неприятно пахнущие гнойные выделения из носа могут быть следствием роста Staphylococcus aureus или Pseudomonas aeruginosa. Влияние носительства S. aureus на патогенез, течение и частоту развития рецидивов ГПА долгие годы было предметом активного изучения [21-23]. С.В. Клименко в 2006 году подтвердила, что носительство S. aureus в полости носа ассоциировано с достоверным увеличением частоты рецидивов ГПА [24]. В 2010 году Laudien и соавт. показали, что частота носительства S. aureus среди пациентов с ГПА (72%) была несколько выше, чем в группе больных ревматоидным артритом (46%) и среди здоровых сотрудников клиники, на базе которой проходило исследование (58%). При этом риск рецидивов ГПА был выше у носителей стафилококка [25]. Однако при анализе микробиоты носа методом секвенирования 16s рРНК было установлено, что носительство стафилококков у пациентов с ГПА встречается не чаще, чем у здоровых людей [26]. Кроме того, негативное влияние носительства S. aureus на частоту рецидивов ГПА остается спорным. Например, по данным E. Besada и соавт., у пациентов, получавших иммунодепрессанты, как носительство, так и элиминация S. aureus не оказывали никакого влияния на частоту как обострений ГПА, так и гипогаммаглобулинемии [19].
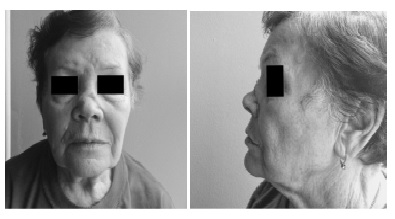
При высокой активности ГПА с поражением ВДП слизистая оболочка носа покрыта диффузными кровоизлияниями, появляются гнойные выделения и корки, что вызывает дополнительное затруднение носового дыхания. Однако симптомы воспаления слизистой оболочки могут быть и менее выраженными, в таких случаях на активность процесса может указывать боль в области носа [18]. Прогрессирование патологического процесса приводит к вовлечению не только слизистой оболочки, но и костно-хрящевого скелета носа. При этом хрящевые структуры наружного носа и перегородки, как правило, поражаются в большей степени, чем костная спинка носа и костная перегородка. Пер форация хрящевой перегородки носа происходит в 33% случаев [16] и может вызывать потерю ее поддерживающей функции. Преимущественно поражается передняя часть носовой перегородки [18], поскольку в ней расположено сосудистое сплетение, обеспечивающее кровоснабжение хряща. В некоторых случаях отек слизистой оболочки и образование корок настолько сильно выражены, что перфорацию носовой перегородки удается визуализировать только после достижения ремиссии, регресса воспалительных изменений и уменьшения объема мягких тканей [27]. Дальнейшее прогрессирование заболевания приводит к увеличению числа перфораций передней части носовой перегородки [28], что влечет за собой значительное разрушение хрящевого скелета: деформации варьируются от потери высоты спинки носа до уменьшения ее длины с нарушением проекции кончика носа и последующим втягиванием носогубного угла. Потеря поддержки кончика приводит к укорочению носа с характерной седловидной деформацией, которая развивается у 23% пациентов с ГПА (рис. 1) [16]. Одновременно с развивающейся седловидной деформацией кожа носа может приобретать синеватую окраску, что связано с нарушением кровоснабжения этой области [29].
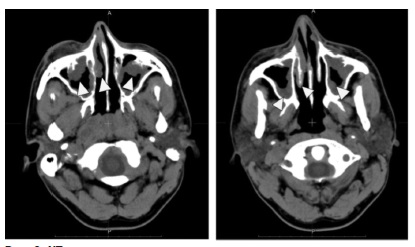
B. D’Anza и соавт. выделили основные варианты поражения ВДП при ГПА, которые могут быть визуализированы при компьютерной (КТ) и магнитно-резонансной (МРТ) томографии: утолщение слизистой оболочки (87,7%), костная деструкция (59,9%) и эрозии носовой перегородки (59,4%) [30]. В острую фазу воспаления диагностическое значение поражения придаточных пазух носа в рамках ГПА может быть несколько переоценено, поскольку КТ и МРТ не позволяют дифференцировать гранулематозное воспаление при васкулите от инфекционного гранулематозного воспаления. В хронической же стадии заболевания, особенно после нескольких рецидивов, околоносовые пазухи постепенно заполняются рубцовой тканью, а верхнечелюстные пазухи часто уменьшаются в объеме, что нередко сочетается с прогрессирующей костной деструкцией и может быть легко обнаружено при помощи КТ и МРТ [31].
Поражение гортани при ГПА наиболее часто проявляется в виде подскладочного стеноза. Это потенциально жизнеугрожающее состояние развивается у 15-25% пациентов с ГПА [6]. При гистологическом исследовании выявляют неспецифический фиброз и инфильтрацию тканей гортани нейтрофилами и эпителиоидными гистиоцитами без явных признаков васкулита [32]. Помимо гортани, в патологический процесс часто вовлекаются бронхи и реже – трахея. Симптомы стеноза трахеи развиваются постепенно и включают в себя сухой кашель, осиплость голоса и одышку. Безусловно, следует учитывать и другие причины возникновения этих жалоб: дифференциальная диагностика проводится между врожденными, травматическими, инфекционными и опухолевыми заболеваниями, ни одно из которых, однако, не сопровождается системными проявлениями, характерными для ААВ.
Микроскопический полиангиит. МПА преимущественно поражает мелкие сосуды почек и легких и не сопровождается развитием гранулематозного воспаления или эозинофилии [33]. По данным Е.М. Щеголевой и соавт., поражение почек наблюдалось у 93,1% больных МПА, легких – у 78,4% [34]. Причиной смерти в 65% случаев становится диффузное альвеолярное кровотечение и/или прогрессирование почечной недостаточности до терминальной стадии, а в 18% – инфекционные осложнения [35]. Среди пациентов с МПА, обследованных в клинике им. Е.М. Тареева, диффузное альвеолярное кровотечение, было зафиксировано в 30,4% случаев, а быстропрогрессирующий гломерулонефрит – в 53% [34].
Распространенность МПА, как и ГПА, также значительно отличается в зависимости от региона. Например, в Японии она составляет 18,2 случаев на миллион населения в год, а Великобритании – 6,5 случаев на миллион населения в год. Средний возраст на момент установления диагноза составил 69 лет в Японии и 60 лет в Великобритании [36], а в России был ниже – 48-51 год [34,37].
Поражение носа и придаточных пазух у пациентов с МПА встречается реже, чем у пациентов с ГПА или ЭГПА, тем не менее, в большинстве исследований частота вовлечения ВДП у больных МПА была достаточно высокой. По данным зарубежных авторов она варьировалась от 9% до 30% [11,62,65], а России составляла от 13,7% по данным Е.М. Щеголевой и соавт. [34] до 54% в исследовании Т.В. Бекетовой [37]. В отличие от ГПА, при МПА не формируются очаги костной деструкции, а при биопсии слизистой оболочки отсутствует гранулематозное воспаление [36].
Несмотря на довольно высокую частоту, подробный анализ различных проявлений поражения ВДП при МПА в литературе практически не представлен. В недавно опубликованном исследовании [3] вовлечение ВДП было выявлено у 57% пациентов с МПА, в том числе изменения придаточных пазух – у 52%, хронический риносинусит – у 26%, рецидивирующие носовые кровотечения – у 26%, перфорация носовой перегородки – у 8,7%. Следует отметить, что если диагноз МПА не был морфологически верифицирован, наличие выраженных проявлений со стороны ВДП у таких пациентов указывает на необходимость пересмотра диагноза в пользу возможного ГПА.
Эозинофильный гранулематоз с полиангиитом. ЭГПА характеризуется наличием эозинофильного и некротизирующего гранулематозного воспаления, затрагивающего в том числе дыхательные пути, некротизирующего васкулита, преимущественно поражающего сосуды малого и, реже, среднего калибра, и бронхиальной астмы с эозинофилией [1]. Ежегодная заболеваемость ЭГПА оценивается в 0,5-4,2 случаев на миллион населения в год, а распространенность составляет 11-14 случаев на миллион населения [38-40].
Приблизительно у половины пациентов с ЭГПА наблюдается аллергический ринит с затруднением носового дыхания и хроническим полипозным риносинуситом (рис. 2) [41]. A. Bacciu и соавт. наблюдали полипоз носа и аллергический ринит у 76,1% и 42,8% из 28 больных ЭГПА, соответственно, а хронический риносинусит в анамнезе – у 14,2% [42]. В другом исследовании частота полипоза носа у 29 пациентов с ЭГПА составила 58,6% [43]. Среди 93 пациентов с ЭГПА, обследованных в клинике им. Е.М. Тареева, частота поражения ЛОР-органов, в том числе ринита, синусита или полипоза носа, достигла 88,2% случаев и не зависела от наличия АНЦА [44].
A. Cottin и соавт. [45] выявили хронический риносинусит у 114 (73%) из 157 пациентов с ЭГПА, в том числе аллергический – у 21% и неаллергического типа – у 52%. При КТ признаки синусита имелись у 66% пациентов, однако образование корок в носовых ходах при осмотре было выявлено только у 15% пациентов. Полипоз носа был диагностирован у 53% больных, причем более половины из них нуждались в оперативном лечении, а частота возникновения рецидивов после оперативного вмешательства составила 50%. В исследовании Y. Nakamaru и соавт. хронический риносинусит был диагностирован на ранних стадиях ЭГПА более чем у 80% пациентов [46]. При проведении цитологического исследования мазка со слизистой оболочки носа у больных ЭГПА с симптомами хронического риносинусита V. Seccia и соавт. обнаружили гиперэозинофилию в 17 из 28 случаев [47]. При гистологическом исследовании обычно выявляют некротическое гранулематозное воспаление в сочетании с тканевой эозинофилией [1,48,49].
Полипоз носа при ЭГПА нередко оказывается устойчивым к лечению глюкокортикостероидами и часто рецидивирует после хирургического вмешательства, что может потребовать назначения иммунодепрессантов [50]. При диагностике ЭГПА следует учитывать высокую частоту (до 50%) обнаружения риносинусита и полипов полости носа при бронхиальной астме [51].
H. Andersen и соавт. не выявили взаимосвязи между активностью ЭГПА и выраженностью проявлений со стороны ЛОР-органов [52]. Ранее сходные данные были получены V. Seccia и соавт. [47]. По мнению авторов, полученные результаты могут указывать на развитие независимого аутоиммунного воспалительного процесса в области ВДП и органа слуха, подобного локальным формам ГПА, что обосновывает необходимость долгосрочного наблюдения и междисциплинарного подхода к лечению.
Недавно B. Saha и соавт. впервые описали деструктивное поражение ВДП при ЭГПА в виде эрозивного хондрита и седловидной деформации носа [53]. Поражение гортани при ЭГПА практически не описано в литературе, однако еще в одном исследовании V. Seccia и соавт. обнаружили локальные признаки воспаления гортани без формирования подскладочного стеноза у 72% из 43 пациентов с ЭГПА [54]. Тем не менее, авторы склонны связывать высокую частоту вовлечения гортани не с первичным поражением в рамках ЭГПА, а с гастроэзофагеальной рефлюксной болезнью, возникающей вследствие приема глюкокортикостероидов.
Диагностика
При развитии симптомов поражения ВДП в дебюте ААВ, особенно до присоединения системных проявлений, оториноларинголог становится первым специалистом, к которому обращается пациент с системным васкулитом, поэтому от него зависит своевременность постановки диагноза и начала патогенетического лечения заболевания.
Диагностические и классификационные критерии. В настоящее время четкие критерии диагностики ААВ отсутствуют. Общепринятыми по-прежнему остаются классификационные критерии ГПА и ЭГПА, предложенные Американской коллегией ревматологов (АКР) еще в 1990 году. Они были разработаны на основании данных, полученных при обследовании пациентов с предположительным диагнозом системного васкулита [55]. Классификационные критерии МПА в то время не были предложены, поскольку заболевание было выделено в отдельную нозологическую форму только в 1994 г. [56].
В классификационные критерии ГПА входят четыре показателя: воспаление слизистой оболочки носа или рта, патологические изменения по данным рентгенографии грудной клетки, аномальный мочевой осадок и наличие гранулематозного воспаления по данным биопсии. При выявлении двух из четырех признаков заболевание может быть классифицировано как ГПА. Эти критерии обладает чувствительностью 88,2% и специфичностью 92% [57].
В критерии диагноза ЭГПА входят бронхиальная астма, эозинофилия крови >10%, риносинусит, легочные инфильтраты (иногда преходящие), гистологические признаки васкулита с внесосудистым расположением эозинофилов и моно- или полинейропатия [1,5]. При наличии 4 из 6 критериев заболевание может быть классифицировано как ЭГПА с чувствительностью 85% и специфичностью 99,7%.
В 2016 году V. Cottin и соавт. предложили пересмотреть подход к диагностике ЭГПА и классифицировать случаи бронхиальной астмы с гиперэозинофилией более 1500 в мкл при отсутствии признаков васкулита и АНЦА в циркуляции как отдельную нозологическую форму – гиперэозинофильную астму с системными проявлениями (Hypereosinophilic asthma with systemic manifestations, HASM). При этом прежнюю номенклатуру ЭГПА было предложено использовать для тех случаев, когда имеются достоверные проявления или несколько суррогатных критериев васкулита [58].
Классификации критерии МПА отсутствуют, а проявления этого васкулита со стороны ВДП во многом сходны с таковыми ГПА. Это создает определенные трудности, если проведение биопсии носа невозможно или если ее результаты сомнительные. Тем не менее, это заболевание имеет характерные отличия: как уже указано выше, поражение ЛОР-органов при МПА встречается гораздо реже, а локальные формы болезни не описаны, поэтому чаще всего поражение ВДП при МПА сочетается с поражением легких по типу диффузного альвеолярного кровотечения или с поражением почек.
Для установления диагноза МПА принято руководствоваться алгоритмом Европейского агентства по лекарствам (EMA), который основывается на определениях, сформулированных на конференциях в ЧапелХилле [59,60]. В отечественной работе с участием 251 пациента этот алгоритм также был признан эффективным методом определения нозологической принадлежности ААВ [61]. На основании очень крупного международного исследования DCVAS (The Diagnostic and Classification Criteria for Vasculitis) недавно были разработы новые классификационные критерии ААВ, однако они пока не опубликованы.
Роль АНЦА в диагностике и прогнозе ААВ. Наличие в крови АНЦА не входит в “старые" классификационные критерии ААВ. Действительно, существуют так называемые “АНЦА-отрицательные" варианты морфологически доказанного васкулита, при которых выявить эти антитела в крови не удается. Тем не менее, проведение биопсии не всегда бывает доступным и информативным методом диагностики, поэтому наличие АНЦА способствует подтверждению диагноза и рассматривается как дополнительный диагностический признак.
Почти в 90% случаев АНЦА-ассоциированного ГПА определяются антитела к протеиназе-3 (ПР3), в то время как антитела к миелопероксидазе (МПО) присутствует менее чем в 5% случаев [62]. Сходные данные приводит Т.В. Бекетова: у 95% пациентов с ГПА были выявлены антитела к ПР3 и лишь у 7% – антитела к МПО [63]. Интересно, что в китайской популяции наблюдалась обратная картина: антитела к МПО были обнаружены у 70,9% пациентов с ГПА, а к ПР3 – у 29,1% [64].
При МПА частота выявления АНЦА составляет 95%. В 70% случаев определяются антитела к МПО, в остальных – антитела к ПР3. В то же время у пациентов с ЭГПА частота обнаружения АНЦА значительно ниже – примерно 30% (чаще всего к МПО) [58,65]. В исследовании, проводившемся в клинике им. Е.М. Тареева, АНЦА были выявлены у 37 (39,8%) больных ЭГПА, в том числе к МПО – у 34 и к ПР-3 – у 3 [44].
Основным методом типирования и количественного определения АНЦА является иммуноферментный анализ (ИФА). Следует отметить, что предварительное проведение реакции непрямой иммунофлюоресценции перед антиген-специфическим ИФА в настоящее время не является обязательным условием для обеспечения максимальной диагностической точности, что было доказано Европейской группой по изучению васкулитов (EUVAS) [66].
Влияние наличия и типа АНЦА на течение и прогноз заболевания. В настоящее время активно изучается влияние наличия и типа АНЦА на течение и исход заболевания. Например, при ГПА повышенный уровень антител к ПР-3 во время ремиссии ассоциируется с увеличением риска развития рецидивов у пациентов с тяжелым поражением почек или альвеолярным кровотечением в анамнезе, а также у больных, получающих ритуксимаб [67]. В исследовании MAINRITSAN сохранение повышенного уровня АНЦА в крови после 12-месячной поддерживающей терапии было фактором риска развития рецидива ААВ [68]. При МПА повторное повышение титра антител к МПО при условии их предшествующего исчезновения на фоне лечения также ассоциироваось с риском возникновения рецидива [69]. В исследовании С. Durel и соавт. наличие АНЦА у больных ЭГПА коррелировало с более частым поражением почек и нервной системы [70].
Обсуждалась возможность использования АНЦА с целью выделения различных форм ЭГПА – с преимущественно гранулематозным поражением или признаками васкулита [65,71]. Тем не менее, наличие АНЦА недостаточно для выделения таких групп пациентов, так как они присутствуют не более чем у 50% больных ЭГПА, а у одной трети АНЦА-позитивных пациентов не удается выявить прямых или косвенных признаков васкулита [58].
Классификация ААВ в зависимости от серотипа АНЦА. В последние годы некоторые авторы предлагали классифицировать ААВ в соответствии с типом циркулирующих АНЦА, т.е. ПР3-АНЦА или МПО-АНЦА ассо циированный васкулит. В исследовании S. Lionaki и соавт. было доказано, что наличие антител к ПР3 позволяет предсказать более частые рецидивов ААВ. В той же работе было продемонстрировано, что поражение ВДП при наличии ПР3-АНЦА развивается значительно чаще, чем при наличии МПО-АНЦА. Например, деструктивное поражение носового скелета в 94% случаев сопровождалось присутствием антител к ПР3 [72]. Данные клиники им. Е.М. Тареева также свидетельствуют о том, что наличие ПР3-АНЦА ассоциируется с более частым поражением ВДП по сравнению с МПОАНЦА ассоциированными вариантами васкулита [73]. J. Schirmer и соавт. пришли к выводу, что наличие антител к МПО обусловливает более частое формирование подскладочного стеноза и локальной формы ГПА [74], в то время как E. Miloslavsky и соавт. продемонстрировали, что возраст, клинические проявления и риск рецидива больше зависят от типа ААВ, чем от типа циркулирующих антител [75]. Недавно S. Deshayes и соавт. также показали, что наличие антител к ПР3 или к МПО не оказывают существенного влияния на клиническую картину и прогноз ААВ [76].
Тем не менее, генетические исследования методом полногеномного поиска ассоциаций (genome-wide association studies, GWAS) показывают, что с теми или иными генами первично ассоциирован именно тип АНЦА, а не клинические синдромы, на которых основана традиционная классификация ААВ [77]. Это означает, что необходимы дальнейшие многоцентровые исследования, конечной целью которых будет совершенствование формулировки диагноза ААВ для эффективного прогнозирования течения и возникновения рецидивов.
Особенности ранней диагностики поражения ВДП
Симптомы поражения ВДП на ранних стадиях ААВ или при их локальных формах демонстрирует широкую клиническую вариабельность и не обладают специфичностью. В этих случаях спектр дифференциально-диагностического поиска должен включать хронические инфекции (спирохетозы, микобактериозы, аспергиллезы) и другие воспалительные состояния (саркоидоз, другие васкулиты, последствия интраназального употребления кокаина), а также онкологические заболевания (NK/T-клеточная лимфома, назофарингеальная карцинома) [78,79], которые подобно ААВ могут быстро прогрессировать, вовлекая соседние структуры, в том числе ткани орбиты и основание черепа [80]. Раннее подозрение на ААВ позволяет ускорить начало лечения и предотвратить развитие системных проявлений заболевания, включая жизнеугрожающее поражение легких и почек. Кроме того, изолированное поражение ВДП в рамках ААВ без адекватной терапии может привести к значительной деструкции костно-хрящевых структур и мягких тканей головы [49].
Исключать ААВ следует у всех пациентов с персистирующими симптомами воспалительного поражения ВДП, не отвечающих на антибактериальную и местную противовоспалительную терапию. В таких случаях необходимо провести общие анализы крови и мочи, биохимическое исследование крови, включая креатинин и С-реактивный белок, компьтерную томографию легких при наличии показаний, а также определить АНЦА. В то же время следует учитывать, что титр АНЦА не коррелирует с тяжестью заболевания [49], а отрицательные результаты теста не исключают наличия васкулита [3]. Гистологическое исследование имеет очень важное значение для дифференциальной диагностики ААВ, однако оно возможно далеко не всегда, а интерпретация полученных данных в значительной степени зависит как от взятого образца ткани, так и от квалификации и опыта патологоанатома. По данным A. Masiak и соавт., только в 32% из из 25 биоптатов слизистой оболочки носа больных ГПА были выявлены изменения, характерные для этого васкулита [81]. K. Devaney и соавт., изучившие 126 биоптатов органов головы и шеи у 70 пациентов с ГПА, также показали невысокую самостоятельную диагностическую ценность гистологического исследования: наличие васкулита, некроза и гранулематозного воспаления одновременно было обнаружено лишь в 16% случаев, а два из указанных признаков – в 23% [82]. Эти данные свидетельствуют о том, что отсутствие АНЦА в сыворотке крови или отрицательные или неоднозначные данные биопсии при подозрении на ААВ могут существенно увеличить время до установления диагноза [83,84].
Наибольшую трудность представляет распознавание изолированных форм ААВ, не сопровождающихся повышенным титром АНЦА в сыворотке крови. По данным A. Knopf и соавт., отсутствие как ПР3-АНЦА, так и МПО-АНЦА в крови является характерной особенностью локального поражения ВДП при ГПА [49]. J. Wojciechowska и соавт. также наблюдали эту тенденцию [3]. Кроме того, обе группы исследователей отмечают более молодой возраст пациентов с серонегативным статусом и локальным поражением ВДП. В этих случаях высокая настороженность ЛОР-врача и, как результат, проведение биопсии имеют ключевое значение для диагностики ААВ и своевременного начала лечения. Интересно, что в противоположной ситуации, т.е. при повышенном титре АНЦА и отсутствии четких признаков поражения ВДП, некоторые исследователи также рекомендуют проведение биопсии макроскопически неизмененной слизистой оболочки носа для подтверждения диагноза [49].
Лечение
Иммуносупрессивная терапия. Подход к терапии ААВ значительно изменился за последние два десятилетия. В настоящее время лечение разделяют на две фазы: индукцию ремиссии, во время которой проводят терапию высокими дозами глюкокортикостероидов (ГКС) в сочетании с сильнодействующими иммуносупрессивными средствами (циклофосфамидом, ритуксимабом и др.), и поддерживающую терапию, когда иммуносупрессивное лечение продолжают для сохранения достигнутой ремиссии. Фаза поддержания ремиссии остается предметом многочисленных клинических исследований, в которых изучают эффективность различных лекарственных препаратов и оптимальную продолжительность лечения, достаточную для успешного предотвращения рецидивов. Для выбора оптимального объема лечения каждому пациенту необходимо проводить оценку активности и тяжести заболевания с помощью Бирмингемской шкалы активности васкулита (Birmingham Vasculitis Activity Score, BVAS) [85,86].
При тяжелых формах заболевания ГКС назначают в дозе 1 мг/кг преднизолона в сутки (не более 60 мг/сут), которую постепенно снижают до поддерживающей. Выбор иммунодепрессанта зависит от тяжести течения заболевания. При поражении жизненно важных органов назначают циклофосфамид внутривенно в режиме “пульс"-терапии, которая более предпочтительна, чем пероральный прием, в связи с более низкой кумуля тивной дозой препарата и меньшим количеством осложнений, связанных с токсическим поражением мочевыводящей системы. В качестве альтернативы циклофосфамиду может быть использован ритуксимаб, который не вызывает бесплодие, что обосновывает его применение у пациентов репродуктивного возраста.
В соответствии с рекомендациями Европейской антиревматической лиги (EULAR)/Европейской почечной ассоциации и Европейской ассоциации диализа и трансплантации (ERA-EDTA) 2015 г. для индукции ремиссии ААВ, не сопровождающего жизнеугрожающими проявлениями, может быть использована комбинация ГКС и метотрексата или микофенолата мофетила [87]. Пациентам с нормальной функцией почек и изолированным поражением ВДП без костных эрозий, разрушения хряща и обонятельной дисфункции, можно назначать метотрексат (20-25 мг/нед перорально или парентерально). По эффективности этот препарат не уступал циклофосфамиду, хотя для достижения ремиссии в некоторых случаях требовалось больше времени, а риск рецидивов был выше [88]. Использование микофенолата мофетила (в дозе 1,5-2 г/сут) имеет меньшую доказательную базу. По данным некоторых исследований, он по эффективности значительно уступает циклофосфамиду [89]. Следует отметить, что время достижения клинической ремиссии при изолированном поражении ВДП и системных формах заболевания по некоторым данным достоверно не отличается [49], что еще раз подчеркивает необходимость не менее серьезного подхода к лечению ограниченных форм ААВ.
Более низкие дозы лекарственных препаратов используются у пожилых людей и пациентов с высоким риском осложнений иммуносупрессивной терапии. При возникновении рецидива и необходимости повторной индукции ремиссии следует принимать во внимание опыт предыдущего лечения, кумулятивную дозу препаратов, степень повреждения органов и риск возникновения инфекций.
Для предотвращения рецидивов после достижения ремиссии назначают поддерживающую терапию. Было доказано, что полная отмена ГКС является самостоятельным предиктором рецидива, поэтому распространенной практикой является применение низких доз ГКС в качестве одного из компонентов поддерживающей терапии ААВ [90].
Согласно рекомендациям EULAR/ERA-EDTA, азатиоприн и метотрексат одинаково эффективны для поддержания ремиссии [87]. Применение ритуксимаба по сравнению с азатиоприном позволяет добиться снижения частоты рецидивов заболевания за 28 месяцев наблюдения при сопоставимой частоте развития тяжелых нежелательных явлений [91]. Микофенолата мофетил менее эффективен для поддержания ремиссии, чем другие препараты, поэтому применяется только в тех случаях, когда альтернативные схемы оказываются неэффективными или не могут быть использованы [92].
Комбинация триметоприма с сульфаметоксазолом в дозе 800/160 мг два раза в день может эффективно снизить риск возникновения рецидивов, особенно у пациентов с изолированным поражением носа и придаточных пазух [93]. Поддерживающую терапию всех ААВ следует продолжать в течение не менее 2 лет после достижения ремиссии [87].
Приведенных общих принципов индукционной и поддерживающей иммуносупрессивной терапии рекомендуется придерживаться при всех трех нозологических формах ААВ. Однако в отношении лечения ЭГПА уровень доказанности и степень рекомендаций ниже, чем для других ААВ. Пациентам, не соответствующим критериям тяжелого течения заболевания, внутривенное введение циклофосфамида не рекомендовано. Кроме того, при ЭГПА единственным иммуносупрессивным препаратом, рекомендуемым для поддержания ремиссии, является азатиоприн [87]. Оценка эффективности ритуксимаба для индукции ремиссии у пациентов с ЭГПА в настоящее время проводится Французской группой по исследованию васкулитов в исследовании REOVAS. Недавно для лечения ЭГПА было одобрено применение меполизумаба – моноклональных антител к интерлейкину-5 [94].
Хирургическое лечение. При позднем начале патогенетической терапии ГПА прогрессирующая деструкция костной и хрящевой ткани наружного носа может приводить к появлению седловидной деформации носа, которая является заметным косметическим дефектом и может оказывать значительное влияние на качество жизни больных. Тем не менее, до настоящего времени остается распространенной позиция отказа от выполнения хирургических вмешательств у пациентов с ГПА [18,95,96]. Выбор консервативной тактики ведения нередко продиктован опасениями, что хирургическое лечение может привести к обострению как локальных, так и системных проявлений ГПА. Однако эта настороженность основана на анализе лишь отдельных клинических наблюдений. A. Coordes и соавт. обобщили 41 случай хирургического лечения деформаций наружного носа и перфорации носовой перегородки у пациентов с ГПА со средним периодом наблюдения 2,6 года [28]. В большинстве случаев использовались аутотрансплантаты костей черепа или реберных хрящей, однако в некоторых исследованиях применялись аллотрансплантаты. По данным анализа, хирургическое лечение седловидной деформации носа у пациентов с ГПА при отсутствии или минимальной активности локального воспаления не было сопряжено с более высокой частотой послеоперационных осложнений (инфекций раны, смещением или резорбцией трансплантата) по сравнению с таковой при подобных операциях у пациентов, не страдающих ГПА. Тем не менее, реконструкция наружного носа при ГПА ассоциировалась с некоторым увеличением частоты ревизионных операций. Некото рые исследователи отмечают, что результаты оперативных вмешательств при локальной форме ГПА в целом лучше, чем при вовлечении в патологический процесс нескольких систем органов [97].
При поражения гортани под голосовыми складками и трахеи стратегия ведения пациентов заключается в использовании трех компонентов лечения: системная иммуносупрессивная терапия, антибактериальные и антигрибковые препараты, а также местная терапия [98]. К последней относятся баллонная дилатация, лазерная терапия и криотерапия, аргоноплазменная коагуляция, диатермокоагуляция, местное использование ГКС, митомицина С и алемтузумаба (моноклонального антитела к антигену лимфоцитов CD52) [33].
Когда хронический воспалительный процесс в стенках гортани приводит к формированию стойкого стеноза, хирургическое лечение может быть необходимым, чтобы избежать установки постоянной трахеостомы [99]. Если стеноз локализован в области перстневидного хряща, могут быть использованы различные методики трансплантации тканей для ларингопластики [18,100]. C. Costantino и соавт. описали 11 случаев ларинготрахеальной резекции и реконструкции у пациентов с ГПА с последующим наблюдением в течение 10 лет [32]. В 55% случаев понадобилось проведение дополнительных дилатаций после хирургического лечения стеноза, при этом лишь в одном случае была установлена постоянная трахеостома. Высокая вероятность развития стеноза нижних дыхательных путей (18%) обосновывает проведение регулярного и, как правило, пожизненного контроля состояния после проведенной операции.
В целом объективно оценить эффективность и безопасность хирургических вмешательств при ААВ достаточно сложно. Тем не менее, иногда их проведение необходимо, а наиболее значимыми факторами, позволяющими снизить число осложнений, являются минимизация местного воспаления тканей в оперируемой области и дополнительного повреждения тканей в результате процедуры [80].
Местная терапия. Использование топических лекар ственных препаратов для лечения поражений слизистой оболочки ВДП при ААВ заслуживает отдельного внимания вследствие своей простоты и эффективности. В исследовании B. Hofauer и соавт. было отмечено, что среди 20 пациентов с вовлечением ВДП в рамках ААВ, несмотря на проведение адекватной иммуносупрессивной терапии, в 61,1% случаев отмечалось лишь незначительное уменьшение симптомов поражения ВДП или отсутствие положительной динамики [48]. Разрушение реснитчатого эпителия носа изменяет транспорт слизи и способствует персистированию хронической инфекции в носовых ходах и образованию слизисто-гнойных корок, которые могут быть устранены с помощью промывания носа различными солевыми растворами в сочетании с применением назальных мазей для смягчения корок [18]. B. Hofauer и соавт. предложили применять локальную липосомальную терапию с использованием спреев с фосфолипидами, которые стабилизируют липидный слой и поддерживают естественную защитную функцию слизистой оболочки носа. Использование этого метода в течение двух месяцев позволило значительно уменьшить выраженность симптомов со стороны ВДП, причем как по стандартизованным оценочным шкалам BVAS, так и по данным анкетирования пациентов [48].
Влияния поражения ВДП на прогноз заболевания
В 1996 году L. Guillevin и соавт. разработали шкалу (Five-Factor Score, FFS), предназначенную для прогнозирования течения васкулита на основании числа органов и систем, вовлеченных в патологический процесс [101]. Первоначальная версия этой шкалы была предназначена для оценки трех некротизирующих васкулитов – узелкового полиартериита, МПА и ЭГПА. В 2009 году FFS была пересмотрена, что сделало возможным ее применение при трех типах ААВ, включая ГПА. По данным нового исследования, поражение ВДП и органа слуха при ГПА, даже при генерализованном варианте болезни, ассоциировалось с более низким относительным риском смерти, а их отсутствие – наоборот, ухудшало общий прогноз [102].
В 2016 году C. Durel и соавт. в многоцентровом исследовании у пациентов с ЭГПА подтвердили, что поражение ВДП является не только предиктором лучшей выживаемости, но и связано с меньшей частотой поражения почек и сердца в рамках системного аутоиммунного процесса [70]. Недавно M. Felicetti и соавт. получили сходные данные у пациентов с ГПА: поражение ВДП было ассоциировано с более высокими показателями пятилетней выживаемости, а также служило независимым предиктором благоприятного исхода заболевания [103].
Таким образом, поражение ВДП не только является одним из ключевых симптомов АНЦА-ассоциированных васкулитов, но и, по мнению некоторых авторов, может указывать на более благоприятное течение ААВ и более низкий риск смерти независимо от наличия или отсутствия АНЦА.
| ГПА | МПА | ЭГПА | |
|---|---|---|---|
| Характер поражения ВДП | Геморрагические и гнойно-геморрагические корки в носовых ходах, носовые кровотечения, хронический риносинусит, перфорация носовой перегородки, седловидная деформация носа, подскладочный стеноз гортани |
Хронический риносинусит, носовые кровотечения | Хронический риносинусит, полипоз носа |
| Гистологические особенности | Гранулематозное воспаление | Васкулит сосудов мелкого калибра без гранулематозного воспаления | Гранулематозное воспаление, тканевая эозинофилия |
| Преобладающий тип АНЦА | ПР3-АНЦА, реже МПО-АНЦА | МПО-АНЦА, реже ПР3-АНЦА | МПО-АНЦА (редко) |
Основные различия между ААВ в отношении характерных вариантов поражения ВДП, гистологической картины и преобладающего серотипа АНЦА были объединены нами в сводную (табл. 1.)
Заключение
Поражение ВДП часто встречается при ААВ и в течение длительного времени может оставаться единственным симптомом заболевания. Хотя выживаемость пациентов с ААВ определяется в основном вовлечением легких и почек, а поражение ВДП ассоциировано с более благоприятным прогнозом, хроническое воспаление в ЛОРорганах может привести к необратимому повреждению лицевого скелета и значительно ухудшает качество жизни больных, а вовлечение в патологический процесс гортани и трахеи прямо связано с риском развития жизнеугрожающих осложнений. Ранняя диагностика васкулита и своевременное начало иммуносупрессивной терапии позволяют избежать необратимых изменений ЛОР-органов и существенно улучшить прогноз заболевания в целом. В связи с этим особое значение приобретает информированность и настороженность в отношении ААВ врачей различных специальностей, в первую очередь оториноларингологов.
Используемые источники
- Jennette JC, Falk R, Bacon P, et al. 2012 revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum 2013;65:1-11. Pearson T, Bremmer M, Cohen J, Driscoll M. Vasculopathy related to cocaine adulterated with Levamisole: A review of the literature. Dermatol Online J 2012;18(7):1.
- Wojciechowska J, KręCicki T. Clinical characteristics of patients with granulomatosis with polyangiitis and microscopic polyangiitis in ENT practice: a comparative analysis. Acta Otorhinolaryngol Ital 2018;38(6):517-27.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992;116:488-98.
- Fries JF, Hunder GG, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis: summary. Arthritis Rheum 1990;33:1135-6.
- Langford CA, Sneller MC, Hallahan CW, et al. Clinical features and therapeutic management of subglottic stenosis in patients with Wegener’s granulomatosis. Arthritis Rheum 1996;39:1754-60.
- Новиков П.И., Моисеев С.В., Кузнецова Е.И. и др. Изменения течения заболевания и прогноза гранулематоза с полиангиитом (Вегенера): результаты 40-летнего наблюдения. Клин фармакол тер 2014;1:32-7. [Novikov PI, Moiseev SV, Kuznetsova EI, et al. Changes in the clinical course and prognosis of granulomatosis with polyangiitis (Wegener’s) over 40 years. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2014;1:32-6 (In Russ.)].
- Pearce FA, Grainge MJ, Lanyon PC, et al. The incidence, prevalence and mortality of granulomatosis with polyangiitis in the UK Clinical Practice Research Datalink. Rheumatology 2017;56:58996.
- O’Donnell JL, Stevanovic VR, Frampton C, et al. Wegener’s granulomatosis in New Zealand: evidence for a latitude-dependent incidence gradient. Intern Med J 2007;37:242-6.
- Naidu GSRSNK, Misra DP, Rathi M, Sharma A. Is granulomatosis with polyangiitis in Asia different from the West? Int J Rheum Dis 2019;22 Suppl 1:90-4.
- Wojciechowska J, Krajewski W, Krajewski P, Krecicki T. Granulomatosis with polyangiitis in otolaryngologist practice: a review of current knowledge. Clin Exp Otorhinolaryngol 2016;9:8-13.
- Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener’s granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med 1983;98:76-85.
- Hughes RG, Drake-Lee A. Nasal manifestations of granulomatous disease. Hosp Med 2001;62(7):417-21.
- Gottschlich S, Ambrosch P, Kramkowski D, et al. Head and neck manifestations of Wegener's granulomatosis. Rhinology 2006;44(4):227-33.
- Report of the Rhinosinusitis Task Force Committee Meeting. Alexandria, Virginia, August 17, 1996. Otolaryngol Head Neck Surg 1997;117(3 Pt 2):S1-68.
- Cannady SB, Batra PS, Koening C, et al. Sinonasal Wegener granulomatosis: a single-institution experience with 120 cases. Laryngoscope 2009;119(4):757-61.
- Morales-Angulo C, García-Zornoza R, Obeso-Agüera S, et al. Ear, nose and throat manifestations of Wegener’s granulomatosis (granulomatosis with polyangiitis). Acta Otorrinolaringologica (English Ed) 2012;63:206-11.
- Rasmussen N. Management of the ear, nose, and throat manifestations of Wegener granulomatosis: an otorhinolaryngologist's perspective. Curr Opin Rheumatol 2001;13(1):3-11.
- Besada E, Koldingsnes W, Nossent JC. Staphylococcus aureus carriage and longterm rituximab treatment for granulomatosis with polyangiitis. Peer J 2015;3:e1051.
- Wong RJ, Gliklich RE, Rubin PA, Goodman M. Bilateral nasolacrimal duct obstruction managed with endoscopic techniques. Arch Otolaryngol Head Neck Surg 1998;124:703-6.
- Pinching AJ, Rees AJ, Pussell BA, et al. Relapses in Wegener's granulomatosis: the role of infection. BMJ 1980;281:836-838.
- Stegeman CA, Cohen Tervaert JW, Sluiter WJ, et al. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med 1994;120:12-7.
- Popa ER, Stegeman CA, Van der Meer B, et al. Staphylococcus toxic-shocksyndrome toxin-1 (TSST-1): a risk factor for disease exacerbation in Wegener's granulomatosis. Cleveland Clin J Med 2002;69(suppl II):27.
- Клименко С.В. Гранулематоз Вегенера: клинические особенности современного течения, прогностические факторы, исходы. М., 2006, 119 с. [Klimenko SV. Wegener’s granulomatosis: clinical manifestations, prognostic factors and outcomes. Moscow, 2006, 119 p. (In Russ.)].
- Laudien M, Gadola SD, Podschun R, et al. Nasal carriage of Staphylococcus aureus and endonasal activity in Wegener’s granulomatosis as compared to rheumatoid arthritis and chronic rhinosinusitis with nasal polyps. Clin Exper Rheumatol 2010;28(suppl.57):51-5.
- Rhee RL, Sreih A, Grayson PC, et al. Nasal microbiota in patients with granulomatosis with polyangiitis compared to healthy controls. 2017 ACR/ARHP Annual Meeting: Abstract number 2935.
- Trimarchi M, Gregorini G, Facchetti F, et al. Cocaine-induced midline destructive lesions. Medicine 2001;80:391-404.
- Kridel RW. Considerations in the etiology, treatment, and repair of septal perforations. Facial Plast Surg Clin North Am 2004;12(4):435-50.
- Coordes A, Loose SM, Hofmann VM, et al. Saddle nose deformity and septal perforation in granulomatosis with polyangiitis. Clin Otolaryngol 201;43(1):291-9.
- D'Anza B, Langford CA, Sindwani R. Sinonasal imaging findings in granulomatosis with polyangiitis (Wegener granulomatosis): A systematic review. Am J Rhinol Allergy 2017;31(1):16-21.
- Muhle C, Reinhold-Keller E, Richter C, et al. MRI of the nasal cavity, the paranasal sinuses and orbits in Wegener's granulomatosis. Eur Radiol 1997;7:566-70.
- Costantino CL, Niles JL, Wright CD, et al. Subglottic stenosis in granulomatosis with polyangiitis: the role of laryngotracheal resection. Ann Thorac Surg 2018;105(1):249-53.
- Nasser M, Cottin V. The respiratory system in autoimmune vascular diseases. Respiration 2018;96:12-28.
- Щеголева Е.М., Буланов Н.М., Виноградова Е.С. и др. Варианты течения и исходы микроскопического полиангиита. Клин фармакол тер 2018;27(3):35-
- [Shchegoleva EM, Bulanov NM, Vinogradova ES, et al. Clinical variants and outcomes of microscopic polyangiitis. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2014;1:32-6 (In Russ.)].
- Краева В.В., Бекетова Т.В. Легочное кровотечение в практике ревматолога. Терапевтический архив 2019;91(5):76-83. [Kraeva VV, Beketova TV. Lung hemorrhage in the practice of rheumatologist. Terapevticheskii Arkhiv 2019;91(5): 76-83 (in Russ.)].
- Fujimoto S, Watts RA, Kobayashi S, et al: Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the UK. Rheumatology 2011;50:1916-1920.
- Бекетова Т.В. Микроскопический полиангиит, ассоциированный с антинейтрофильными цитоплазматическими антителами: особенности клинического течения. Терапевтический архив 2015;87(5):33-46. [Beketova TV. Microscopic polyangiitis associated with ANCA: clinical manifestations. Terapevticheskii Arkhiv 2015;87(5):33-46 (In Russ.)].
- Mohammad A, Jacobsson L, Mahr A, et al. Prevalence of Wegener’s granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology 2007;46:1329-37.
- Watts RA, Lane S, Scott DG. What is known about the epidemiology of the vasculitides? Best Pract Res Clin Rheumatol 2005;19:191-207.
- Mahr A, Guillevin L, Poissonnet M, Aymé S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Care Res 2004;51:92-9.
- Hofauer B, Chaker A, Thürmel K, Knopf A. Manifestations of autoimmune disorders in otorhinolaryngology. HNO 2017;65(8):695-708
- Bacciu A, Bacciu S, Mercante G, et al. Ear, nose and throat manifestations of Churg-Strauss syndrome. Acta Otolaryngol 2006;126:503-9.
- Bacciu A, Buzio C, Giordano D, et al. Nasal polyposis in Churg-Strauss syndrome. Laryngoscope 2008;118:325-9.
- Загвоздкина Е.С., Новиков П.И., Моисеев С.В. Особенности клинических проявлений и течения эозинофильного гранулематоза с полиангиитом в зависимости от наличия антител к цитоплазме нейтрофилов. Клин фармакол тер 2017;26(1):24-30. [Zagvozdkina ES, Novikov PI, Moiseev SV. Clinical features of eosinophilic granulomatosis with polyangiitis in ANCA-positive and ANCA-negative patients. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2017;26(1):24-30 (In Russ.)].
- Cottin V, Bel E, Bottero P, et al. Respiratory manifestations of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Eur Respir J 2016;48:1429-41.
- Nakamaru Y, Takagi D, Suzuki M, et al. Otologic and rhinologic manifestations of eosinophilic granulomatosis with polyangiitis. Audiol Neurootol 2016;21:45-53.
- Seccia V, Baldini C, Latorre M, et al. Focus on the involvement of the nose and paranasal sinuses in eosinophilic granulomatosis with polyangiitis (Churg-Strauss Syndrome): Nasal cytology reveals infiltration of eosinophils as a very common feature. Int Arch Allergy Immunol 2018;175(1-2):61-9.
- Hofauer B, Bas M, Strassen U, et al. Liposomal local therapy of sinunasal symptoms in ANCA associated vasculitis. Laryngorhinootologie 2014;93:461-66.
- Knopf A, Chaker A, Stark T, et al. Clinical aspects of granulomatosis with polyangiitis affecting the head and neck. Eur Arch Otorhinolaryngol 2015;272:185-93.
- Brescia G, Zanotti C, Parrino D, et al. Nasal polyposis pathophysiology: Endotype and phenotype open issues. Am J Otolaryngol 2018;39(4):441-4.
- Porsbjerg C, Menzies‐Gow A. Comorbidities in severe asthma: Clinical impact and management. Respirology 2017;22:651-661.
- Andersen H, Götz P, Bremer JP, et al. Manifestation of eosinophilic granulomatosis with polyangiitis in the head and neck area over time taking systemic disease activity into consideration. Z Rheumatol 2018;77:928.
- Saha B, Saha A, Cordeiro-Rudnisky F, et al. Destructive upper airway disease from eosinophilic granulomatosis with polyangiitis (EGPA): the very first case. Case Rep Rheumatol 2019:May 23;1-4.
- Seccia V, Cristofani-Mencacci L, Dallan I, et al. Eosinophilic granulomatosis with polyangiitis and laryngeal involvement: review of the literature and a crosssectional prospective experience. J Laryngol Otol 2018;1-5.
- Rao JK, Allen NB, Pincus T. Limitations of the 1990 American College of Rheumatology classification criteria in the diagnosis of vasculitis. Ann Intern Med 1998;129:345-52.
- Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: The proposal of an international consensus conference. Arthritis Rheum 1994;37:187-92.
- Bloch DA, Michel BA, Hunder GG, et al. The American College of Rheumatology 1990 criteria for the classification of vasculitis: patients and methods. Arthritis Rheum 1990;33:1068-73.
- Cottin V, Bel E, Bottero P, et al. Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): a study of 157 patients by the Groupe d’Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun Rev 2017;16:1-9.
- Watts R, Lane S, Hanslik T, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. An Rheum Dis 2006;66(2):222-7.
- Abdulkader R, Lane SE, Scott DGI, Watts RA. Classification of vasculitis: EMA classification using CHCC 2012 definitions. Ann Rheum Dis 2013;72(11):1882-8.
- Бекетова Т.В. Алгоритм диагностики системных васкулитов, ассоциированных с антинейтрофильными цитоплазматическими антителами. Терапевти ческий архив 2018;90(5):13-21. [Beketova TV. Diagnostic algorythm for anti-neutrophil cytoplasmic antibody associated systemic vasculitis. Terapev ticheskii Arkhiv 2018;90(5)13-21 (In Russ.)].
- Basu N, Watts R, Bajema I, et al. EULAR points to consider in the development of classification and diagnostic criteria in systemic vasculitis. Ann Rheum Dis 2010;69:1744-50.
- Бекетова Т.В. Гранулематоз с полиангиитом, патогенетически связанный с антинейтрофильными цитоплазматическими антителами: особенности клинического течения. Научно-практическая ревматология 2012;6(50):19-28. [Beketova TV. Granulomatosis with polyangiitis, which is pathogenetically associated with antineutrophil cytoplasmic antibodies: clinical features. Rheumatology Science and Practice 2012;50(6):19-28. (In Russ.)].
- Chang DY, Li ZY, Chen M, Zhao MH. Myeloperoxidase-ANCA-positive granulomatosis with polyangiitis is a distinct subset of ANCA-associated vasculitis: A retrospective analysis of 455 patients from a single center in China. Semin Arthritis Rheum 2019;48(4):701-6.
- Comarmond C, Pagnoux C, Khellaf M, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum 2013;65:270-81.
- Damoiseaux J, Csernok E, Rasmussen N, et al. Detection of antineutrophil cytoplasmic antibodies (ANCAs): a multicentre European Vasculitis Study Group (EUVAS) evaluation of the value of indirect immunofluorescence (IIF) versus antigen-specific immunoassays. Ann Rheum Dis 2016;76(4):647-53.
- Fussner LA, Hummel AM, Schroeder DR, et al. Factors determining the clinical utility of serial measurements of antineutrophil cytoplasmic antibodies targeting proteinase 3. Arthr Rheumatol 2016;68:1700-10.
- Terrier B, Pagnoux C, Geri G, et al. Factors predictive of ANCA-associated vasculitis relapse in patients given Rituximab-maintenance therapy. Arthritis Rheum 2014;66(Suppl10):779.
- Terrier B, Saadoun D, Sène D, et al: Antimyeloperoxidase antibodies are a useful marker of disease activity in antineutrophil cytoplasmic antibody-associated vasculitides. Ann Rheum Dis 2009;68:1564-71.
- Durel C-A, Berthiller J, Caboni S, et al. Long-term follow up of a multicenter cohort of 101 patients with eosinophilic granulomatosis with polyangiitis (ChurgStrauss). Arthritis Care Res 2016;68(3):374-87.
- Sable-Fourtassou R, Cohen P, Mahr A, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005;143:632-8.
- Lionaki S, Blyth ER, Hogan SL et al. Classification of antineutrophil cytoplasmic autoantibody vasculitides: The role of antineutrophil cytoplasmic autoantibody specificity for myeloperoxidase or proteinase 3 in disease recognition and prognosis. Arthr Rheum 2012;64(10):3452-62.
- Буланов Н.М., Макаров Е.А., Шеголева Е.М. и др. Взаимосвязь антительного профиля и клинического течения поражения почек при АНЦА-ассоциированных васкулитах. Терапевтический архив 2018;90(6):15-21. [Bulanov NM, Makarov EA, Shchegoleva EM, et al. Relationship between serological profile (ANCA type) and clinical features of renal involvement in ANCA-associated vasculitides. Terapev ticheskii Arkhiv 2018;90(6)15-21 (In Russ.)].
- Schirmer JH, Wright MN, Herrmann K, et al. Myeloperoxidase–antineutrophil cytoplasmic antibody (ANCA)-positive granulomatosis with polyangiitis (Wegener's) is a clinically distinct subset of ANCA-associated vasculitis: a retrospective analysis of 315 patients from a German vasculitis referral center. Arthritis Rheumatol 2016;68:2953-63.
- Miloslavsky EM, Lu N, Unizony S, et al. Myeloperoxidase–antineutrophil cytoplasmic antibody (ANCA)-positive and ANCA-negative patients with granulomatosis with polyangiitis (Wegener's): distinct patient subsets. Arthritis Rheumatol 74 2016;68:2945-52.
- Deshayes S, Martin Silva N, Khoy K, et al. Clinical impact of subgrouping ANCA-associated vasculitis according to antibody specificity beyond the clinicopathological classification. Rheumatology (Oxford) 2019;58(10):1731-39.
- Smith K. Genetic studies in ANCA-associated vasculitis point to a new, practical disease classification based on autoantibody specificity. The 18th International Vasculitis and ANCA Workshop, Tokyo; March 2017.
- Fuchs HA, Tanner SB. Granulomatous disorders of the nose and paranasal sinuses. Curr Opin Otolaryngol Head Neck Surg 2009;17:23-7.
- Trimarchi M, Bussi M, Sinico RA, et al. Cocaine-induced midline destructive lesions an autoimmune disease? Autoimmun Rev 2013;12:496-500.
- Trimarchi M, Sinico RA, Teggi R, et al. Otorhinolaryngological manifestations in granulomatosis with polyangiitis (Wegener’s). Autoimmun Rev 2013;12(4):501-5.
- Masiak A, Zdrojewski Z, Pęksa R, et al. The usefulness of histopathological examinations of non-renal biopsies in the diagnosis of granulomatosis with polyangiitis. Rheumatology 2017;55(5):230-6.
- Devaney KO, Travis WD, Hoffman G, et al. Interpretation of head and neck biopsies in Wegenerʼs granulomatosis. Amer J Surg Pathol 1990;14(6):555-64.
- Finkielman JD, Lee AS, Hummel AM, et al. ANCA are detectable in nearly all patients with active severe Wegener’s granulomatosis. Am J Med 2007;120:e9-14.
- Raynaud P, Garrel R, Rigau V, et al. How can the diagnostic value of head and neck biopsies be increased in Wegener’s granulomatosis: a clinicopathologic study of 49 biopsies in 21 patients. Ann Pathol 2005;25(2):87-93.
- Luqmani RA, Bacon PA, Moots RJ, et al. Birmingham Vasculitis Activity Score (BVAS) in systemic necrotizing vasculitis. QJM 1994;87:671-8.
- Mukhtyar C, Lee R, Brown D, et al. Modification and validation of the Bir mingham Vasculitis Activity Score (version 3). Ann Rheum Dis 2009;68:1827-32.
- Yates M, Watts RA, Bajema IM, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis 2016;75(9): 1583-94.
- Faurschou M, Westman K, Rasmussen N et al. Brief Report: long-term outcome of a randomized clinical trial comparing methotrexate to cyclophosphamide for remission induction in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2012;64(10):3472-7.
- Tuin J, Stassen PM, Bogdan DI et al. Mycophenolate mofetil versus cyclophosphamide for the induction of remission in nonlife-threatening relapses of antineutrophil cytoplasmic antibody-associated vasculitis: randomized, controlled trial. Clin J Am Soc Nephrol 2019;14(7):1021-28.
- Walsh M, Merkel PA, Mahr A et al. Effects of duration of glucocorticoid therapy on relapse rate in antineutrophil cytoplasmic antibody-associated vasculitis: a meta-analysis. Arthritis Care Res 2010;62(8):1166-73.
- Guillevin L, Pagnoux C, Karras A, et al: Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N Engl J Med 2014;371:1771-80.
- Hiemstra TF, Walsh M, Mahr A, et al. Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA 2010;304(21):2381-8.
- Stegeman CA, Cohen Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprimsulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. N Engl J Med 1996;335:16-20.
- Faverio P, Bonaiti G, Bini F, et al. Mepolizumab as the first targeted treatment for eosinophilic granulomatosis with polyangiitis: a review of current evidence and potential place in therapy. Ther Clin Risk Manag 2018;Dec7;14:2385-96.
- Drake-Lee AB, Bickerton RC, Milford C. Wegener's granulomatosis and nasal deformity. Br J Clin Pract 1988;42(8):348-50.
- Mann W, Bumb P, Marker-Hermann E. Chronic rhinosinusitis with septal perforation. Differential diagnostic considerations. HNO 2008;56(11):1129-34.
- Congdon D, Sherris DA, Specks U et al. Long-term follow-up of repair of external nasal deformities in patients with Wegener's granulomatosis. Laryngoscope 2002;112(4):731-7.
- Martinez Del Pero M, Jayne D, Chaudhry A, et al. Long-term outcome of airway stenosis in granulomatosis with polyangiitis (Wegener granulomatosis): an observational study. JAMA Otolaryngol Head Neck Surg 2014;140:1038-44.
- Lebovics RS, Hoffman GS, Leavitt RY, et al. The management of subglottic stenosis in patients with Wegener's granulomatosis. Laryngoscope 1992;102:1341-5.
- Prakash UB, Golbin JM, Edell ES, Specks U. Airway involvement in Wegener's granulomatosis. Rheum Dis Clin North Am 2007;33(4):755-75.
- Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996;75:17-28.
- Guillevin L, Pagnoux C, Seror R, et al. French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011;90(1):19-27.
- Felicetti M, Cazzador D, Padoan R, et al. Ear, nose and throat involvement in granulomatosis with polyangiitis: how it presents and how it determines disease severity and long-term outcomes. Clin Rheumatol 2018;37(4):1075-83.