Клинические рекомендации по диагностике и лечению системного амилоидоза
В клинических рекомендациях, подготовленных специалистами различного профиля, рассматриваются методы диагностики и лечениясистемного амилоидоза, в том числе АА (вто-ричный амилоидоз при хронических воспалительных заболеваниях, включая ревматоидныйартрит, анкилозирующий спондилит, аутовоспалительные заболевания, хроническиенагноения, злокачественные опухоли и др.), AL (амилоидоз при плазмоклеточных дискразиях – идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема) иATTR (транстиретиновый; семейные формыполиневропатического, кардиопатического идругого амилоидоза, системный старческийамилоидоз). Диагноз амилоидоза, которыйможно заподозрить на основании клиническихданных, необходимо подтвердить при гистологическом исследовании (окрашивание препаратов ткани конго-красным с последующей микроскопией в поляризованном свете). Чтобы замедлить или приостановить прогрессирование амилоидоза любого типа, необходимо добиться уменьшения количества (или, если возможно, удаления) белков-предшественников путем лечения хронического воспаленияпри АА-амилоидозе или подавления пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуно-глобулинов при AL-амилоидозе. Для замедления прогресирования ATTR-амилоидоза упациентов с полиневропатией применяют тафамидис, который ингибирует диссоциацию мутантного транстиретина и снижает его амилоидогенность.
Определение, классификация, группы риска и принципы диагностики
Амилоидоз – группа заболеваний, отличительным признаком которых является отложение в тканях и органах фибриллярного гликопротеида амилоида. Специфическое свойство амилоида, отличающее его от других фибриллярных белков стромы, – способность к двойному лучепреломлению, что проявляется свечением в поляризованном свете предварительно окрашенных конгокрасным препаратов амилоида с изменением красного цвета конгофильных амилоидных отложений на яблочно-зеленый (дихроизм).
В основе амилоидогенеза лежит синтез большого количества нестабильных белковпредшественников, которые агрегируются с образованием амилоидной фибриллы. Клю чевое значение имеет амилоидогенность основного белка-предшественника амилоида, специфичного для каждой формы амилоидоза (в настоящее время известно более 30 таких белков), обозначение которого положено в основу современной классификации заболевания (ВОЗ, 2016 г.). Названия типов амилоида включают в себя букву А, означающую “амилоид", и обозначение конкретного фибриллярного белка амилоида – А (амилоидный А-протеин), L (легкие цепи иммуноглобулинов), TTR (транстиретин), β2М (β2-микроглобулин), В (В-протеин), IAPP (островковый амилоидный полипептид). Используют также производные наименования – иммуноглобулиновый амилоидоз (AL), транстиретиновый (ATTR) и др. (табл. 1) [1-3]. Следует отметить, что Международная классификация болезней (МКБ) 10-го пересмотра базируется на клиническом принципе, не учитывает особенности патогенеза различных форм амилоидоза и не позволяет обосновать адекватное лечение.
| Белок амилоида | Белок-Белок-предшественник | Клиническая форма амилоидоза |
|---|---|---|
| АА | SSA-белок | Вторичный амилоидоз при хронических воспалительных заболеваниях, в том числе периодической болезни и синдроме Макла-Уэллса |
| AL | λ, κ-легкие цепи иммуноглобулинов | Амилоидоз при плазмоклеточных дискразиях – идиопатический, при миеломной болезни и макроглобулинемии Вальденстрема |
| ATTR | Транстиретин | Семейные формы полиневропатического, кардиопатического и др. амилоидоза, системный старческий амилоидоз |
| Аβ2М | β2-микроглобулин | Диализный амилоидоз |
| AGel | Гелсолин | Финская семейная амилоидная полиневропатия |
| AApoAI | Аполипопротеин А-I | Амилоидная полиневропатия (III тип по van Allen, 1956 г.) |
| AFib | Фибриноген | Амилоидная нефропатия |
| Aβ2 | β-белок | Болезнь Альцгеймера, синдром Дауна, наследственные кровоизлияния в мозг с амилоидозом |
| APrPScr | Прионовый белок | Болезнь Крейтцфельда-Якоба, болезнь Герстманна-Штраусслера-Шейнкера |
| AANF | Предсердный натрийуретический фактор | Изолированный амилоидоз предсердий |
| AIAPP | Амилин | Изолированный амилоидоз в островках Лангерганса при сахарном диабете 2 типа, инсулиноме |
| ACal | Прокальцитонин | При медуллярном раке щитовидной железы |
| ACys | Цистатин С | Наследственные кровоизлияния в мозг с амилоидозом (Исландия) |
АА-амилоидоз чаще всего развивается при ревматоидном артрите, серонегативных спондилоартропатиях, аутовоспалительных наследственных периодических лихорадках, в том числе периодической болезни (семейной средиземноморской лихорадке), а также при хронических нагноениях, туберкулезе. АА-амилоид образуется из сывороточного предшественника SAA (serum amyloid A) – острофазового белка, продуцируемого в значительных количествах в ответ на воспаление. По этой причине АА-амилоидоз называют также реактивным или вторичным.
Клинические формы AL-амилоидоза обусловлены единым этиологическим фактором – В-лимфоцитарной дискразией, характеризующейся формированием аномального клона плазматических или В-клеток в костном мозге, которые продуцируют аномальные иммуноглобулины, обладающие амилоидогенностью (легкие цепи моноклонального иммуноглобулина, чаще λ, реже κ-типа). При первичном AL-амилоидозе плазмоклеточная дискразия относительно более доброкачественная, в то время как при В-гемобластозах (множественной миеломе, болезни Вальденстрема и др.) она обладает признаками злокачественной опухоли. Аномальный амилоидогенный клон плазматических клеток может формироваться также из плазмоцитов, локализующихся вне костного мозга, что может привести к развитию локального амилоидоза. Наиболее распространенные локальные формы AL-амилоидоза – амилоидоз трахеи, бронхов и гортани, мочевого пузыря. Выявление плазмоклеточной дискразии необходимо для диагностики AL-амилоидоза, а также для оценки его риска и дифференциального диагноза.
ATTR-амилоидоз является необратимо прогрессирующим заболеванием с высокой степенью инвалидизации вследствие тяжелого поражения сердца, периферической и/или автономной полиневропатии. Пациенты обычно умирают в течение 10-12 лет от первых проявлений. Развитие ATTR-амилоидоза обусловлено мутациями в молекуле транстиретина или возрастным нарушением секреции тетрамеров транстиретина печенью. В обоих случаях происходит распад тетрамеров транстиретина до мономеров, обладающих выраженной конформационной нестабильностью.
Рекомендации:
- Скрининг АА-амилоидоза следует проводить в следующих группах риска: серопозитивные и серонегативные хронические полиартриты (ревматоидный артрит, анкилозирующий спондилоартрит, ювенильный хронический артрит, псориатический артрит, синдром Рейтера и др.), воспалительные заболевания кишечника (болезнь Крона, язвенный колит), аутовоспалительные заболевания (подагра тяжелого рецидивирующего течения, семейные периодические лихорадки – периодическая болезнь, криопиринопатии, TRAPS, гипериммуно глобулинемия D), хронические нагноения (туберкулез, бронхоэктатическая болезнь, остеомиелит и др.), злокачественные солидные опухоли [1,2,4-6].
- Риск АА-амилоидоза у больных с хроническими воспалительными заболеваниями повышается при персистирующем увеличении уровней маркеров острой фазы воспаления (С-реактивный белок, SAA), наличии анемии хронических заболеваний (с повышением уровня ферритина крови), особенно в сочетании с суставным синдромом (синовитом) [4,6-12]. Дина мический контроль за уровнем этих показателей необходим также при мониторировании течения диагностированного АА-амилоидоза. Для оценки риска развития или прогрессирования АА-амилоидоза в условиях преимущественного аутовоспаления или с целью выявления субклинической активации воспаления можно определять сывороточный маркер нейтрофильной активности S100A12 (кальгранулин) [4,13-15].
- Диагностика аутовоспалительных заболеваний предполагает, в первую очередь, проведение генетического исследования на мутации генов MEFV (пирин), NLRP3 (криопирин), TRAPS (рецептор к фактору некроза опухоли альфа), мевалонаткиназы [4,16-25].
- Высокая частота олигосекреторных моноклональных гаммапатий у лиц старше 50 лет требует скринингового обследования этой группы лиц на предмет моноклональных гаммапатий. Наиболее чувствительным и недорогим турбидиметрическим методом для скрининговой диагностики являет Freelite-метод количественной оценки уровня свободных легких цепей иммуноглобулинов. Все больные с плазмоклеточными дискразиями и лимфопролиферативными заболеваниями входят в группу риска AL-амилоидоза [26-34].
- У пациентов с морфологически подтвержденным амилоидозом диагностика AL-амилоидоза предполагает проведение иммунохимического исследования с применением высокочувствительных методов – иммунофиксации сыворотки и суточной мочи, количественного определения свободных легких цепей иммуноглобулинов (Freelite) [26-35].
- Помимо выявления моноклональной гаммапатии, диагностика плазмоклеточной дискразии предполагает выявление и оценку количества плазмоцитов костного мозга, а также их структурных особенностей. Применение цитогенетического исследования и иммунофенотипирования плазмоцитов важно для уточнения клональности и злокачественности аберрантного клона плазматических клеток, в особенности в редких случаях неинформативности иммунохимического исследования крови и суточной мочи [26,33-34,36,37].
- Важное значение в диагностике плазмоклеточных дискразий имеют также иммуногистохимические методы выявления патологического клона плазматических клеток. Это особенно важно для диагностики, типирования и лечения локального варианта ALамилоидоза [35].
- ATTR-амилоидоз следует подозревать у пациентов с полиневропатией, необъяснимым чередованием запоров и диареи, синдромом карпального канала, особенно при наличии полиневропатии, синдрома карпального канала, кардиомиопатии у родственников. Важными симптомами являются геморрагии, фестончатый край зрачка, потеря массы тела, снижение зрения [38-47].
- Диагностика амилоидоза основывается на результатах морфологического исследования [1-2,39,48].
- При системном амилоидозе для диагностики амилоидного поражения органа нет необходимости проводить его биопсию у больных с ранее верифицированным диагнозом амилоидоза по результатам биопсии другого органа. Однако точная диагностика возможна только с помощью морфологического исследования [1,48].
- С целью выявления амилоида необходимо окрашивание препаратов ткани красителем конго-красный с последующей микроскопией в поляризованном свете. Окончательный диагноз амилоидоза устанавливают при выявлении конгофильных масс, обладающих способностью к яблочно-зеленому или желтоватому свечению в поляризованном свете [1,39,48]. Для более точной диагностики амилоидоза применяют также метод окраски тиофлафином Т, который дает светло-зеленое свечение амилоида [1,2].
- При системном амилоидозе информативна биопсия прямой или двенадцатиперстной кишки (с захватом подслизистого слоя). ATTR-амилоид отличается слабой конгофилией. По этой причине выявить этот тип амилоида нередко удается только при повторных биопсиях из разных органов – аспирационной биопсии подкожной жировой клетчатки, биопсии слюнных желез губ и др. Наиболее эффективна биопсия пораженного органа [1-2,39,48]. У пациентов с синдромом запястного канала исследованию на амилоид необходимо подвергать ткань, удаленную при оперативной декомпрессии запястного канала.
- Не рекомендуется проводить биопсию подкожной жировой клетчатки у больных инсулинозависимым сахарным диабетом, так как в местах иньекций инсулин может агрегировать и формировать амилоидные депозиты [48].
- Для дифференциальной диагностики АА-амилоидоза от AL- и ATTR-амилоидоза используют окрасочные методы при тщательном учете клинических предпосылок разных типов амилоидоза [48,49].
- Наиболее эффективным методом типирования является иммуногистохимическое исследование. Поскольку некоторые антисыворотки могут давать перекрестные реакции с разными типами амилоида, исследование целесообразно проводить с панелью антисывороток. Для неспециализированных терапевтических и нефрологических стационаров рекомендуется применение панели антисывороток к SAA, разным типам тяжелых цепей иммуноглобулинов, легким цепям иммуноглобулинов λ и κ, транстиретину. Важно также использовать антисыворотки к фибриногену [36,50].
- Для диагностики ATTR-амилоидоза необходимо генетическое исследование на наличие мутации гена транстиретина [39].
Клинические проявления
Для вторичного АА-амилоидоза характерно более раннее начало, чем для AL-амилоидоза (средний возраст больных составляет около 40 и 65 лет, соответственно). ATTR-амилоидоз, несмотря на наследственную природу, характеризуется низкой пенетрантностью и также проявляется обычно после 35 лет.
Поражение почек – ведущий клинический признак АА- и AL-амилоидоза, наблюдающийся практически у всех больных. Поражение почек встречается и у больных с многими формами семейного амилоидоза (AFib, ALys, AGel и др.). При ATTR-амилоидозе нефропатия отмечается лишь у 20-23% больных. Клинически амилоидная нефропатия характеризуется неуклонно прогрессирующим течением с последовательной сменой стадий: протеинурия, нефротический синдром, хроническая почечная недостаточность (ХПН). Иногда возможно развитие ХПН без предшествующего нефротического синдрома.
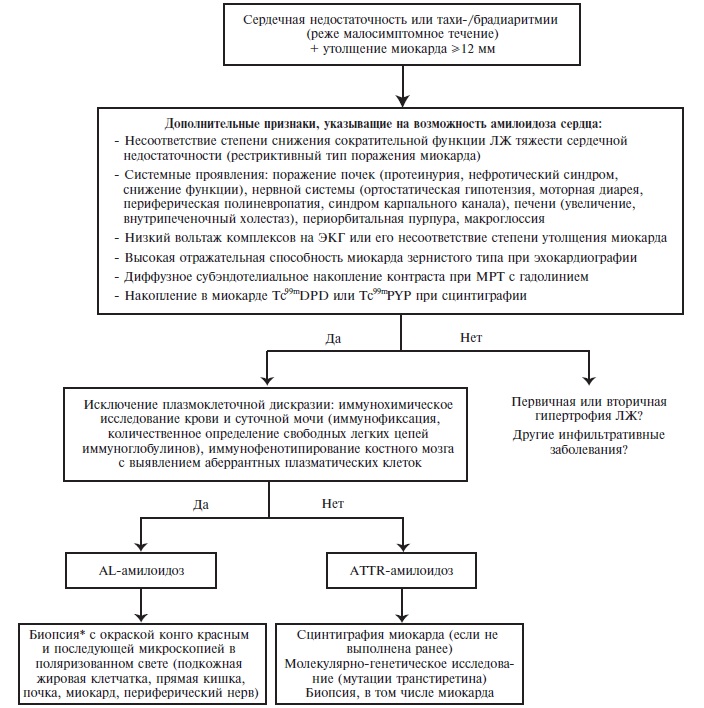
Поражение сердца развивается у подавляющего большинства больных AL-амилоидозом и у 50-60% пациентов с АTTR-амилоидозом, но не характерно для АА-амилоидоза (рис. 1). При эхокардиографии у больных амилоидозом сердца наблюдается утолщение межжелудочковой перегородки и стенки левого желудочка (чаще симметричное), которое не сопровождается электрокардиографическими признаками гипертрофии миокарда. У части больных отмечается снижение вольтажа зубцов на ЭКГ, хотя отсутствие этого признака не исключает диагноз амилоидоза сердца. Нарушение диастолической функции левого желудочка (рестриктивный тип) приводит к развитию сердечной недостаточности, которая быстро прогрессирует, плохо поддается лечению и почти у 50% пациентов оказывается причиной смерти. Кроме того, у больных амилоидозом сердца часто наблюдаются различные аритмии и нарушения проводимости.
При AL-амилоидозе и особенно ATTR-амилоидозе часто встречается ортостатическая артериальная гипотензия – вариант сосудистой недостаточности, при которой сосуды теряют способность поддерживать нормальное артериальное давление в условиях ортостатических нагрузок. Она проявляется ощущением дурноты и потемнением в глазах в ортостазе в сочетании с резким снижением АД. Обычно этот симптом связан с дисфункцией автономной нервной системы (амилоидоз нервных сплетений сосудов). Тяжелая ортостатическая гипотензия сопровождается обмороками, а иногда приводит к развитию острого нарушения мозгового кровообращения.
Поражение желудочно-кишечного тракта может проявляться, особенно при AL-амилоидозе, тяжелой диареей или динамической непроходимостью, которые чаще связаны с нарушениями моторики кишечника вследствие дисфункции автономных нервных сплетений. Иногда выявляют изъязвления или перфорацию стенок с возможным кровотечением. При поражении пищевода возможна дисфагия.
Поражение печени при АА- и AL-типах амилоидоза наблюдают практически в 100% случаев. Функция печени чаще остается сохранной, редким признаком амилоидоза печени является внутрипеченочная портальная гипертензия. При некоторых вариантах семейного ALys-амилоидоза описаны тяжелые спонтанные внутрипеченочные кровотечения.
Увеличение селезенки, обусловленное амилоидным поражением, отмечается у большинства больных и обычно сопутствует увеличению печени.
Поражение нервной системы, представленное симптомами периферической соматической и автономной невропатии, отмечают у 17-35% больных AL-амилоидозом и практически у всех пациентов с наследственной амилоидной полиневропатией разных типов (ATTR, AApoA1 и др.). В большинстве случаев развивается дистальная симметричная полиневропатия с неуклонно прогрессирующим течением, различные дисфункции автономной нервной системы. Реже выявляют двусторонний синдром запястного канала, обусловленный сдавлением срединного нерва депозитами амилоида.
Поражение кожи наблюдают почти у 40% больных AL-амилоидозом. Помимо параорбитальных геморрагий описаны также папулы, бляшки, узелки, пузырьковые высыпания, склеродермоподобная индурация кожи.
Амилоидные отложения в мышцах чаще встречаются при AL-амилоидозе. Макроглоссия – патогномоничный симптом AL-амилоидоза, развивающийся примерно у 20% пациентов.
Редким проявлением амилоидоза, описанным при AL- и, в особенности, АTTR-типах, бывает поражение глаз (сухой кератоконъюнктивит, вторичная глаукома, помутнение стекловидного тела, дисфункции зрачка).
Клиническая картина других типов амилоидоза варьируется в зависимости от основной локализации и распространенности амилоидных депозитов, которые иногда могут быть значительными и напоминать проявления AL-амилоидоза.
Рекомендации:
- Наиболее типичное проявление амилоидоза почек – изолированная протеинурия более 0,5 г/сут, чаще нефротического уровня. Иногда при множественной миеломе важное значение приобретает иммунохимическое электрофоретическое исследование мочи для отличия альбуминурии в рамках амилоидоза и протеинурии переполнения (наличие в моче белка БенсДжонса, реакция термопреципитации белка Бенс-Джонса не обладает достаточной информативностью). Для установления связи протеинурии с амилоидозом необходимо также исключить протеинурию, связанную с диабетической нефропатией и гипертонической почкой [4,5,29,48,51-56].
- На амилоидоз сердца указывает утолщение межжелудочковой перегородки и/или задней стенки левого желудочка более 12 мм при эхокардиографии, особенно в сочетании с низкоамплитудной ЭКГ. Дифференциальный диагноз проводят с гипертрофией левого желудочка, которая может быть следствием артериальной гипертонии, аортальных пороков, гипертрофической кардиомиопатии и других причин [29,37-39,42,48,51-53,57-62].
- Характерное проявление амилоидоза сердца – низкая амплитуда желудочковых комплексов на ЭКГ (менее 5 мм в отведениях от конечностей). Пато логические Q-зубцы у больных амилоидозом нередко являются псевдоинфарктными (вследствие электрически нейтральных отложений амилоида, имитирующих рубцовые изменения), однако при амилоидозе коронарных артерий возможно развитие и истинного инфаркта миокарда [29,37-39,42,48,51-53,56-62].
- При амилоидозе сердца наблюдается рестриктивное нарушение диастолической функции левого желудочка, фракция выброса часто остается нормальной, или степень ее снижения не соответствует тяжести сердечной недостаточности [29,37-39,42,48,51-53,5662].
- Всем больным с амилоидозом сердца необходимо проведение стандартной эхокардиографии с допплерометрической оценкой трансмитрального крово тока, при наличии технической возможности оправдано также проведение тканевой допплерометрии миокарда для более точной оценки внутрисердечной гемодинамики. В стандарт обследования больных амилоидозом сердца входят также ЭКГ и суточное мониторирование АД и ЭКГ [1,2,29,37-39,42,48,5153,56-62].
- Магнитно-резонансная томография (МРТ) с контрастированием гадолинием с высокой вероятностью выявляет инфильтративный характер поражения миокарда и имеет особое диагностическое значение при “изолированном" поражении сердца [48,56,6062].
- Амилоидную инфильтрацию сердца позволяет выявить также сцинтиграфия миокарда с пирофосфатом технеция, особенно при наличии трудностей в морфологической диагностике ATTR-амилоидоза, для которого характерна слабая конгофилия пораженных тканей. Интенсивное накопление радиоактивного препарата в миокарде (2+/3+) в сочетании с утолщением миокарда неясной этиологии указывает на высоко вероятный ATTR-амилоидоз, если у пациента исключен диагноз AL-амилоидоза (рис. 1) [39,42,58-59,62].
- Диагноз амилоидоза сердца может быть подтвержден при биопсии миокарда. Однако проведение этого исследования обычно не требуется при наличии типичных эхокардиографических изменений у пациентов с амилоидозом, установленным при биопсии другого органа, например, почки или слизистой оболочки прямой или двенадцатиперстной кишки.
- Диагностика периферической амилоидной полиневропатии основывается на клинической оценке неврологических проявлений: обычно выявляют различные нарушения чувствительности, в частности температурной и болевой. Из-за поражения преимущественно мелких немиелинизированных волокон электромиография и исследование скорости проведения нервного импульса обычно неинформативны для ранней диагностики амилоидной полиневропатии [38,39]. Доминирование жалоб, связанных с поражением нервной системы, является отличительной чертой ATTR-амилоидоза.
- Электромиография наряду с другими нейрофизиологическими методами (количественное сенсорное тестирование, конфокальная микроскопия нервов роговицы, оценка состояния интраэпидермальных нервных волокон в биоптате кожи) может использоваться для оценки характера (признаки аксонального и/или аксонально-демиелинизирующего типа поражения двигательных и чувствительных волокон нервов конечностей) и тяжести неврологических нарушений. Электромиография в сочетании с ультразвуковым исследованием периферических нервов позволяет объективизировать их повреждение в анатомически узких каналах (туннельные невропатии) [38,39].
- Признаками I стадии амилоидной полиневропатии являются незначительные нарушения чувствительности по полиневропатическому типу, более выраженные в ногах, пациент сохраняет способность к самостоятельной ходьбе. При II стадии полиневропатии из-за нарастания чувствительных и присоединения двигательных нарушений в виде нижнего периферического преимущественно дистального парапареза походка пациента нарушается, требуется опора на трость или костыли. На III стадии тяжелые двигательные нарушения приковывают пациента к постели, передвижение возможно только на инвалидной коляске [38,39,46].
- Поражение вегетативной нервной системы чаще всего проявляется ортостатической гипотензией разной степени тяжести. Однако систолическое артериальное давление менее 90 мм рт. ст. может быть обусловлено низким сердечным выбросом у больных с сердечной недостаточностью или гиповолемией при тяжелом нефротическом синдроме. Другие частые проявления поражения вегетативной нервной системы – моторная диарея, дисфункция мочевого пузыря, половой сферы, гипогидроз [1,2,38,51]. Наряду с моторной диареей причиной значительного (на 9-18 кг) снижения массы тела могут быть нарушения трофики мышц [38,39].
- Двусторонний синдром запястного канала наиболее характерен для ATTR-, β2М- и AL-амилоидоза и проявляется интенсивными болями и парестезиями в IIII пальцах кистей с постепенной атрофией мышц тенара [38,39].
- МРТ головного мозга с контрастным усилением проводят при наличии клинических признаков поражения центральной нервной системы [38,39].
- Основным признаком амилоидоза печени является ее увеличение, наиболее специфична гепатомегалия более 15 см по данным компьютерной томографии. У больных амилоидозом печени обычно выявляют также холестаз (повышение активности щелочной фосфатазы и/или g-глютамилтранспептидазы в 1,5 раза по сравнению с верхней границей нормы). Ложная диагностика амилоидоза печени возможна у больных с тяжелой застойной правожелудочковой недостаточностью [2,4,48,51].
- Для выявления амилоидоза печени, селезенки и почек всем пациентам проводят ультразвуковое исследование этих органов, в некоторых случаях необходимо проведение компьютерной томографии брюшной полости [2,48].
- Диарея вследствие инфильтрации амилоидом стенки желудочно-кишечного тракта возникает редко, такую диарею трудно дифференцировать от моторной диареи в рамках поражения вегетативной нервной системы. Наиболее надежно вовлечение желудочнокишечного тракта при амилоидозе устанавливают по результатам морфологического исследования. Одна ко обнаружение амилоида только в стенках сосудов желудочно-кишечного тракте не является критерием его поражения, необходимо обнаружение амилоидных депозитов в интерстиции подслизистого слоя кишечника [1-2,5,48,52].
- Нодулярный легочный и трахеобронхиальный амилоидоз за редким исключением – это проявление локального AL-амилоидоза. Для системного AL-амилоидоза характерно обнаружение диффузного интерстициального легочного амилоидоза. В связи с редкостью дыхательной недостаточности необходимости в морфологической верификации легочного амилоидоза обычно не возникает. Наиболее информативным методом диагностики амилоидоза легких является компьютерная томография [1,2,48]. У больных локальным трахеобронхиальным AL-амилоидозом важными методами мониторирования течения заболевания являются ларингоскопия и бронхоскопия [27,48].
- На амилоидоз плевры указывает рецидивирующий плевральный выпот, который не зависит от эффективности лечения отечного синдрома, обусловленного сердечной недостаточностью или нефротическим синдромом. При амилоидном поражении плевры жидкость, полученная во время пункции плевральной полости, нередко содержит примесь крови. При амилоидозе плевры эвакуация плеврального выпота, как правило, малоэффективна из-за быстрого его накопления [1,2,48].
- Поражение мягких тканей характерно для AL-амилоидоза. Макроглоссия с инфильтрацией дна ротовой полости и периорбитальная пурпура (и кожные геморрагии на теле) патогномоничны для этого типа амилоидоза. Возможны также псевдогипертрофия скелетных мышц с развитием мышечной слабости, лимфаденопатия, амилоидоз височной артерии [1,2,39,48,52].
- Признаками прогрессирования амилоидоза сердца являются дальнейшее утолщение миокарда (на 2 мм и более), увеличение функционального класса сердечной недостаточности, снижение фракции выброса левого желудочка на 10% и более. Показатели тяжести амилоидоза сердца – увеличение уровня NTproBNP (особенно более 1800 нг/л) и тропонинов (тропонин Т более 0,025 нг/мл). Критериями прогрессирования амилоидоза почек считают увеличение протеинурии (на 50% от исходного уровня, как правило на 1 г/сут и более) и сывороточного уровня креатинина (на 25% и более от исходного). Информативным критерием прогрессирования амилоидоза печени является увеличение активности щелочной фосфатазы на 50% от исходной. Прогрес сирование полиневропатии объективизируют на основании результатов стимуляционной электромиографии и других нейрофизиологических и нейровизуализационных методов обследования (см. выше). Важным показателем тяжести больных AL-амилоидозом является разница в содержании свободных легких цепей иммуноглобулинов более 180 мг/л, установленная методом Freelite [2,38,51,48,60,61,64].
Лечение системного амилоидоза
Целью терапии любого типа амилоидоза служит уменьшение количества (или, если возможно, удаление) белков-предшественников для того, чтобы замедлить или приостановить прогрессирование болезни. Неблаго приятный прогноз при естественном течении амилоидоза оправдывает применение агрессивных методов лечения. Клиническое улучшение, достигаемое с помощью лечения, включает стабилизацию или восстановление функции жизненно важных органов, а также предотвращение функциональных нарушений с увеличением продолжительности жизни больных. Лечение амилоидоза должно включать симптоматические методы, направленные на уменьшение выраженности сердечной недостаточности, аритмии, отечного синдрома, коррекцию артериальной гипотензии и др.
Лечение АА-амилоидоза
Цель терапии АА-амилоидоза – подавление продукции белка-предшественника SAA (вплоть до устойчивой нормализации), что достигается активным лечением хронического воспаления (в том числе субклинического). Это позволяет уменьшить клинические проявления и предотвратить прогрессирование амилоидной нефропатии и существенно улучшить прогноз.
Рекомендации:
- Основной стратегией лечения АА-амилоидоза является эффективное подавление воспаления. Лечение должно проводиться вне зависимости от клинической активности воспалительного заболевания до нормализации уровня маркеров острой фазы воспаления – С-реактивного белка (предпочтительно применение высокочувствительного метода измерения) и/или SAA [10-13,63,66-77].
- Больным ревматоидным артритом и серонегативными спондилоартропатиями необходима постоянная пожизненная базисная терапия. Предпочтение отдают генно-инженерным биологическим препаратам, в том числе ингибиторам фактора некроза опухоли-α, интерлейкина-6, ритуксимабу. После оценки эффективности и безопасности базисной терапии оправдано присоединение терапии колхицином в дозе 2 мг/сут [10-13,63,66-77].
- Препаратом выбора при периодической болезни и тяжелой рецидивирующей подагре является колхицин в дозе 2 мг/сут. Начальная доза препарата составляет 0,5 мг/сут, затем ее постепенно увеличивают до целевой под контролем клинического анализа крови и сывороточного уровня креатинина. Для предупреждения осмотической диареи, обусловленной колхицином, возможно временное назначение ферментных препаратов. Дозу колхицина снижают до 1 мг/сут у больных с хронической болезнью почек 4-5 стадии [1,2,4,69]. При неэффективности колхицина показано назначение ингибиторов интерлейкина-1, в частности канакинумаба.
- При криопиринопатиях, TRAPS препаратами выбора являются ингибиторы интерлейкина-1, к которым присоединяют колхицин [18,19,74-77].
- При хронических нагноениях важное значение имеет хирургическое лечение. В дальнейшем или одновременно проводят лечение димексидом (5-10 г/сут в разведении большим количеством соков – томатного, гранатового и др.). Препарат предпочтителен при легочных нагноениях [1-2,4,6,31].
Лечение АL-амилоидоза
При AL-амилоидозе, как и при множественной миеломе, целью лечения служит подавление пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуноглобулинов. В отличие от множественной миеломы, принципиальной задачей лечения AL-амилоидоза является по возможности полная элиминация патологического клона. В связи с быстрым прогрессированием заболевания важное значение имеет применение быстродействующих схем лечения на основе бортезомиба. По мере достижения ремиссии у некоторых больных применяют высокодозную химиотерапию с поддержкой аутологичными стволовыми клетками. При строгом подборе больных с исключением противопоказаний к этой терапии эффект достигают у 60% больных. У больных с клиническими симптомами амилоидоза сердца, ортостатической гипотензией, диареей, желудочно-кишечными кровотечениями в анамнезе, а также у лиц старше 70 лет с амилоидным поражением двух и более систем организма проведение высокодозной химиотерапии не рекомендуется. Тяжелый агранулоцитоз и другие осложнения существенно ограничивают ее применение. Проводят также лечение талидомидом или леналидомидом. Колхицин при AL-амилоидозе не эффективен.
Рекомендации:
- Основной стратегией лечения АL-амилоидоза является элиминация амилоидогенного клона плазматических клеток костного мозга. После достижения гематологической ремиссии проводят противорецидивное лечение в течение не менее 12 месяцев. В этот период при отсутствии противопоказаний возможна высокодозная химиотерапия с поддержкой аутологичными стволовыми клетками, которая позволяет достичь длительной ремиссии [2,26,37,56,57,65,78,79].
- Полный гематологический ответ диагностируют на основании исчезновения моноклональных амилоидогенных иммуноглобулинов по данным иммунофиксации крови и суточной мочи, количественного определения свободных легких цепей иммуноглобулинов методом Freelite (нормальный уровень легких цепей κ – менее 19,4 мг/л, λ – менее 26,3 мг/л, нормальное соотношение легких цепей в пределах 0,261,65). При снижении уровня свободных легких цепей на 50% от исходного диагностируют частичный ответ, критерием очень хорошего частичного ответа является разница между содержанием двух типов легких цепей иммуноглобулинов менее 40 мг/л [37,60,61,64].
- Клинический эффект терапии в первую очередь оценивают по динамике кардиологических и ренальных показателей. Особо выделяют NT-proBNP-ответ (снижение уровня маркера на 30% и более или на 300 нг/л и более у пациентов с исходным уровнем более 650 нг/л). С клиническим эффектом лечения больше коррелирует почечный ответ (снижение протеинурии на 75% и более, повышение сывороточного креатинина не более 25% от исходного). Ответ со стороны других органов не обладает существенной прогностической информативностью. Эффективность лечения амилоидоза печени оценивают по снижению активности щелочной фосфатазы (на 50% и более) и уменьшению размеров печени (краниокаудальный размер по данным КТ должен уменьшиться на 30% в течение года после достижения гематологической ремиссии). Эффективность лечения амилоидной полиневропатии определяют, главным образом, по результатам клинического неврологического осмот ра. Уменьшение амилоидных депозитов в мягких тканях может быть оценено по данным компьютерной томографии или МРТ. Эффективным методом оценки общего содержания амилоида в тканях служит сцинтиграфия с радиоактивным амилоидным Ркомпонентом [2,26,37,57,60,61,64].
- Учитывая возможность достижения быстрого гематологического ответа, терапией первой линии, особенно у больных с высоким риском быстрого прогрессирования, считают комбинированные схемы на основе бортезомиба, например, трехкомпонентная схема: бортезомиб 1,3 мг/м2 внутривенно или подкожно (1, 5, 8 и 11-й дни цикла), мелфалан 0,15 мг/кг внутрь (с 1-го по 4-й день) и дексаметазон 20 мг/сут внутрь (1, 5, 8 и 11-й дни). Для большей безопасности предпочтительно подкожное введение бортезомиба. Если в дальнейшем планируется высокодозная химиотерапия с поддержкой аутологичными стволовыми клетками мелфалан в составе трехкомпонентной схемы заменяют на циклофосфамид (400 мг внутривенно капельно в 1, 8, 12-й дни), который не вызывает истощение пула стволовых клеток в костном мозге. Курсы химиотерапии на основе бортезомиба проводят каждые 4 недели (всего 8 курсов). Одновременно назначают омепразол, низкомолекулярные гепарины (для профилактики тромбозов при применении высоких доз дексаметазона), при наличии показаний – антибиотики, противогрибковые препараты, ацикловир [65,78,79].
- Не менее эффективна схема терапии мелфаланом (внутрь 0,15 мг/кг с 1-го по 4-й день) и дексаметазоном (внутрь 20 мг/сут в дни 1-4, 9-12 и 17-21) каждые 4-6 недель. Основным недостатком этой схемы является медленное формирование гематологического ответа, что делает ее менее перспективной у больных с высоким риском быстрого прогрессирования амилоидоза. Одновременно назначают омепразол, низкомолекулярные гепарины (для профилактики тромбозов при применении высоких доз дексаметазона), при наличии показаний – антибиотики, противогрибковые препараты, ацикловир [1,2,37,56,57,65,78,79].
- У тяжелых больных с декомпенсированной сердечной недостаточностью может применяться схема терапии мелфаланом (внутрь 0,15 мг/кг с 1-го по 4-й дни) и преднизолоном (0,8 мг/кг с 1-го по 7-й день). Однако эффективность этой схемы ограничена [1,2,56,57,65,78,79].
Лечение ATTR-амилоидоза
До недавнего времени единственным методом лечения ATTR-амилоидоза была трансплантация печени, секретирующей нормальный транстиретин. Поскольку 98% всего сывороточного транстиретина синтезируется печенью, это позволяло прервать продукцию мутантного транстиретина. Трансплантация печени существенно замедляет прогрессирование ATTR-амилоидоза, а 20летняя выживаемость больных после трансплантации составляла 55,3% [36]. Однако уже имеющиеся массы амилоида способны выступать в роли ядра нуклеации для новых депозитов амилоида на основе нормального транстиретина (амилоидускоряющая субстанция). В настоящее время у больных с ранними стадиями ATTRамилоидной полиневропатии апробированы консервативные методы стабилизации тетрамерной структуры мутантного транстиретина и, следовательно, подавления его амилоидогенности. Один из таких препаратов – тафамидис замедлял на 52% (р=0,027) прогрессиро вание неврологических нарушений у больных ATTRамилоидозом, сохраняя функцию периферических соматических и автономных нервных волокон [12]. В клиническом исследовании III фазы лечение тафамидисом по сравнению с плацебо у больных с ATTR-амилоидозом сердца вызывало снижение общей смертности и частоты госпитализаций по сердечно-сосудистым причинам и задерживало ухудшение функциональной активности [81]. В Российской Федерации применение тафамидиса зарегистрировано только для лечения ATTR-амилоидоза с периферической полиневропатией.
В качестве стабилизатора транстиретина изучается также дифлюнизал из группы нестероидных противовоспалительных препаратов, однако его эффективность показана только в экспериментальных условиях.
Рекомендации:
- Трансплантация печени остается одним из методов лечения ATTR-амилоидоза. Наилучших показателей выживаемости удается достичь у больных моложе 50 лет с небольшой длительностью заболевания, нормальным индексом массы тела и мутацией Val30Met. Эффективность трансплантации печени существенно ниже у больных с III стадией полиневропатии, ортостатической гипотензией, хронической сердечной недостаточностью, удлинением комплекса QRS бо лее 120 мс и утолщением межжелудочковой перегородки [2,38-40,43-46].
- Больным ATTR-амилоидозом с периферической невропатией рекомендуется пожизненный прием тафамидиса в дозе 20 мг/сут внутрь, который обладает хорошим профилем безопасности [38-40,43-46,80].
Заместительная почечная терапия
Рекомендации:
- Поскольку ХПН служит одной из основных причин смерти больных системным амилоидозом, проведение гемодиализа или постоянного амбулаторного перитонеального диализа позволяет улучшить прогноз этих пациентов. Выживаемость больных амилоидозом при проведении гемодиализа, независимо от его типа, сопоставима с выживаемостью больных другими системными заболеваниями и сахарным диабетом. При этом хорошую и удовлетворительную реабилитацию отмечают у 60% пациентов с АА- и AL-типами амилоидоза. Основной причиной смерти больных амилоидозом при проведении гемодиализа являются сердечно-сосудистые осложнения. Амбула торный перитонеальный диализ имеет некоторые преимущества перед гемодиализом, учитывая отсутствие необходимости в постоянном сосудистом доступе и меньший риск артериальной гипотензии во время процедуры диализа. У больных AL-амилоидозом во время процедуры возможно удаление легких цепей иммуноглобулинов [1-4,6,28,48].
- Трансплантация почки эффективна при системном амилоидозе: 5-летняя выживаемость больных и трансплантата составляет 65% и 62%, соответственно, и сопоставима с таковыми в других группах больных с ХПН. Трансплантация почки показана больным с медленным прогрессированием амилоидоза без поражения сердца и желудочно-кишечного тракта. Амилоидоз в трансплантированной почке возникает, по разным данным, примерно у 30% больных, однако он служит причиной потери трансплантата всего у 2-3% пациентов [1-4,6,28,48].
Симптоматическая терапия
Рекомендации:
- Больным амилоидозом противопоказаны сердечные гликозиды и недигидропиридиновые антагонисты кальция (верапамил, дилтиазем), которые могут накапливаться в амилоиде, в то время как β-адреноблокаторы и ингибиторы АПФ следует применять осторожно. Основа лечения застойной сердечной недостаточности при амилоидозе сердца – петлевые диуретики, хотя ортостатическая гипотензия может ограничивать применение этих препаратов, как и β-адреноблокаторов и ингибиторов АПФ [1,2,31,45,49,52,58].
- При наличии желудочковых тахиаритмий, сопровождавющихся высоким риском внезапной сердечной смерти, показана установка искусственного кардиовертера-дефибриллятора, а при синдроме слабости синусового узла и атрио-вентрикулярной блокаде – имплантация искусственного водителя ритма [2,38,39,45,52,53,58].
- Для лечения ортостатической гипотензии могут быть использованы минералокортикостероиды или глюкокортикостероиды, хотя их применение может привести к декомпенсации сердечной недостаточности. Возможно назначение α-адреномиметика мидодрина (Гутрон), однако его применение требует осторожности [1,2,39,42,45,52,58].
- Для контроля кишечной моторики высокую эффективность показали пролонгированные препараты соматостатина (октреотид-лонг 20 мг один раз в месяц) [1,2,38,39,49].
- При деструкции стекловидного тела у больных ATTR-амилоидозом может потребоваться витрэктомия. При наличии неконтролируемой глаукомы проводят трабекулэктомию [39].
- Для купирования невропатической боли у пациентов с амилоидной невропатией рекомендовано использование адъювантных анальгетиков (антиконвульсанты и антидепрессанты) [39].
Используемые источники
- Козловская Л.В., Рамеев В.В., Саркисова И.А. Амилоидоз у пожилых.Клиническая медицина 2005;6:12-20. [Kozlovskaya LV, Rameev VV, SarkisovaIA. Amyloidosis in the elderlies. Klinicheskaya medicina 2005;6:12-20 (In Russ.)].
- Рамеев В.В., Козловская Л.В., Саркисова И.А. Лечение амилоидоза. Врач2007;6:38-41 [Rameev VV, Kozlovskaya LV, Sarkisova IA. Treatment of amyloi-dosis. Vrach 2007;6:38-41 (In Russ.)].
- Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril proteins and amyloido-sis: chemical identification and clinical classification International Society ofAmyloidosis 2016 Nomenclature Guidelines. Amyloid 2016;23(4):209-13.
- Рамеев В.В., Козловская Л.В., Рамеева А.С., Тао П.П. Оптимизация страте-гии ведения больных вторичным АА-амилоидозом. Врач 2019;30(5):3-11[Rameev VV, Kozlovskaya LV, Rameeva AS, Tao PP. Optimization of the man-agement strategy for patients with secondary AA-amyloidosis. Vrach 2019;30(5):3-11 (In Russ.)].
- Gertz MA, Kyle RA. Secondary systemic amyloidosis: response and survival in 64patients. Medicine (Baltimore) 1991;70(4):246-56.
- Lachmann HJ, Goodman HJB, Gilbertson JA, et al. Natural history and outcomein systemic AA amyloidosis. N Engl J Med 2007;356(23):2361–71.
- Kravitz MS, Pitashny M, Shoenfeld Y. Protective molecules – C-reactive protein(CRP), serum amyloid P (SAP), pentraxin3 (PTX3), mannose-binding lectin(MBL), and apolipoprotein A1 (Apo A1), and their autoantibodies: prevalenceand clinical significance in autoimmunity. J Clin Immunol 2005;25(6):582–91.
- Szalai AJ. C-reactive protein (CRP) and autoimmune disease: facts and conjec-tures. Clin Dev Immunol 2004;11(3-4):221–6.
- Volanakis JE. Human C-reactive protein: expression, structure, and function. MolImmunol 2001;38(2-3):189–97.
- Korkmaz C, Ozdogan H, Kasapçopur O, Yazici H. Acute phase response in famil-ial Mediterranean fever. Ann Rheum Dis 2002;61(1):79–81.
- Duzova A, Bakkaloglu A, Besbas N, et al. Role of A-SAA in monitoring subclin-ical inflammation and in colchicine dosage in familial Mediterranean fever. ClinExp Rheumatol 2003;21(4):509–14.
- Lachmann HJ, Sengül B, Yavuzşen TU, et al. Clinical and subclinical inflamma-tion in patients with familial Mediterranean fever and in heterozygous carriers ofMEFV mutations. Rheumatology (Oxford) 2006;45(6):746–50.
- Wittkowski H, Frosch M, Wulffraat N, et al. S100A12 is a novel molecular mark-er differentiating systemic-onset juvenile idiopathic arthritis from other causes offever of unknown origin. Arthritis Rheum 2008;58(12):3924–31.
- Foell D, Wittkowski H, Hammerschmidt I, et al. Monitoring neutrophil activationin juvenile rheumatoid arthritis by S100A12 serum concentrations. ArthritisRheum 2004;50(4):1286–95.
- Kallinich T, Wittkowski H, Keitzer R, et al. Neutrophil-derived S100A12 as novelbiomarker of inflammation in familial Mediterranean fever. Ann Rheum Dis2010;69(4):677–82.
- Benson MD, Liepnieks JJ, Yazaki M, et al. A new human hereditary amyloidosis:the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics2001;72:272–7.
- Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases:concept and clinical manifestations. Clin Immunol Orlando Fla 2013;147(3):155–74.
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokineactivation, and evidence for genetic heterogeneity in patients with neonatal-onsetmultisystem inflammatory disease (NOMID): a new member of the expandingfamily of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002;46(12):3340–8.
- Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisysteminflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med2006;355(6):581–92.
- Touitou I, Lesage S, McDermott M, et al. Infevers: an evolving mutation databasefor auto-inflammatory syndromes. Hum Mutat 2004;24(3):194–8.
- Houten SM, Kuis W, Duran M, et al. Mutations in MVK, encoding mevalonatekinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. NatGenet 1999;22(2):175–7.
- Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduriato hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis 2006;1:13.
- Ruiz Gomez A, Couce ML, Garcia-Villoria J, et al. Clinical, genetic, and thera-peutic diversity in 2 patients with severe mevalonate kinase deficiency. Pediatrics2012;129(2):535-9.
- Bader-Meunier B, Florkin B, Sibilia J, et al. Mevalonate kinase deficiency: a sur-vey of 50 patients. Pediatrics 2011;128(1):152-9.
- Gershoni-Baruch R, Brik R, Zacks N, et al. The contribution of genotypes at theMEFV and SAA1 loci to amyloidosis and disease severity in patients with familialMediterranean fever. Arthritis Rheum 2003;48(4):1149–55.
- Мрыхин Н.Н., Лысенко Л.В., Чеботарева Н.В. и др. Частота выявления иварианты моноклональной гаммапатии у больных многопрофильного тера-певтического стационара. Врач 2019;30(2):54-60 [Mrykhin NN, Lysenko LV,Chebotareva NV, et al. Frequency of detection and variants of monoclonalgammapathy in patients of a multidisciplinary therapeutic hospital. Vrach2019;30(2):54-60 (In Russ.)].
- Тао П.П., Рамеев В.В., Рамеева А.С. и др. Проблемы диагностики и лечениялокального AL-амилоидоза. Клин фармакол тер 2019;28(3):26-33 [Tao PP,Rameev VV, Rameeva AS, et al. Problems of diagnosis and treatment of local AL-amyloidosis. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2019;29(3):26-33. (In Russ.)].
- Gillmore JD, Hawkins PN, Pepys MB. Amyloidosis: a review of recent diagnosticand therapeutic developments. Br J Haematol 1997;99:245-56.
- Leung N, Glavey SV, Kumar S, et al. A detailed evaluation of the current renalresponse criteria in AL amyloidosis: is it time for a revision? Haematologica2013;98(6):988-92
- Bradwell AR. Serum free light chain analysis. 4th edition The Binding Site Ltd.2006;285 p.
- Kyle RA. Amyloidosis: a convoluted story. Br J Haematol 2001;114:529–38.
- Kyle RA. Monoclonal gammopathy of undetermined significance: natural historyin 241 cases. Am J Med 1978;64:814-26.
- Gertz MA, Rajkumar SV. Multiple Myeloma. Diagnosis and Treatment. NewYork:Springer 2014;311 p.
- Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Groupmolecular classification of multiple myeloma: spotlight review. Leukemia 2009;23(12):2210–21.
- Owen-Casey MP, Sim R, Cook HT, et al. Value of antibodies to free light chainsin immunoperoxidase studies of renal biopsies. J Clin Pathol 2014;67:661–6.
- Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloido-sis as AL (primary) amyloidosis. N Engl J Med 2002;346:1786-91.
- Muchtar E, Buadi F, Dispenzieri A, Gertz M. Immunoglobulin light-chain amy-loidosis: from basics to new developments in diagnosis, prognosis and therapy.Acta Haematol 2016;135:172-90.
- Рамеев В.В., Мясников Р.П., Виноградов П.П. и др. Системный ATTR-ами-лоидоз, редкая форма поражения внутренних органов. Рациональная фар-макотерапия в кардиологии 2019;15(3):349-58. [Rameev VV, Myasnikov RP,Vinogradov PP, et al. Systemic ATTR-amyloidosis, a rare form of internal organdamage. Rational pharmacotherapy in cardiology 2019;15(3):349-58 (In Russ.)].
- Adams D, Suhr O, Hund E, et al. First European consensus for diagnosis,manegement, and treatment of transthyretin familial amyloid polyneuropathy/ Curr Opin Neurol 2016;29(suppl 1):14-26.
- Гудкова А.Я., Полякова А.А., Амелин А.В. и др. Не VAL30MET-транстире-тиновая амилоидная кардиомиопатия. Обзор сведений литературы и кли-ническое наблюдение. Российский кардиологический журнал 2018;2(154):121-128 [Gudkova AJ, Polyakova AA, et al. Non-Val30Met-trans thyretin amyloidcardiomyopathy. Review of literature, data and clinical case. Rossijskij kardio-logicheskij zhurnal 2018;2(154):121-8 (In Russ.)].
- Hemminki K, Li X, Försti A, et al. Incidence of hereditary amyloidosis andautoinflammatory diseases in Sweden: endemic and imported diseases. BMC MedGenet 2013;14:88.
- González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-typetransthyretin amyloidosis as a cause of heart failure with preserved ejection frac-tion. Eur Heart J 2015;36:2585–94.
- Adams D, Théaudin M, Cauquil C, et al. FAP neuropathy and emerging treat-ments. Curr Neurol Neurosci Rep 2014;14(3):435.
- Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecularpathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry2015;86(9):1036-43.
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditaryamyloidosis for clinicians. Orphanet J Rare Dis 2013;20(8):31.
- Adams D, Cauquil C, Theaudin M, et al. Current and future treatment of amyloidneuropathies. Expert Rev Neurother 2014;14(12):1437-51.
- Koike H, Tanaka F, Hashimoto R, et al. Natural history of transthyretinVal30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry 2012;83(2):152-8.
- Gertz MA, Comenzo R, Falk RH, et al. Definition of organ involvement andtreatment response in immunoglobulin light chain amyloidosis (AL): a consensusopinion from the 10th International Symposium on Amyloid and Amyloidosis,Tours, France, 18-22 April 2004. Am J Hematol 2005;79(4):319-28.
- Hans LA, Nienhuis JB, Hazenberg BPC. The prevalence and management of sys-temic amyloidosis in western countries. Kidney Dis 2016;2:10-9.
- Benson MD, Liepnieks J, Uemichi T, et al. Hereditary renal amyloidosis associ-ated with a mutant fibrinogen alpha-chain. Nature Gen 1993;3:252–6.
- Rameeva A, Vedanova K, Rameev V, et al. The critical role of chronic kidney dis-ease in the progression of AL-amyloid cardiopathy. Amyloid 2019;26(S1):109-10
- Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory fea-tures in 474 cases. Semin Hematol 1995;32(1):45-59.
- Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary sys-temic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood1992;79:1817–22.
- Pinney JH, Smith CJ, Taube JB, et al. Systemic amyloidosis in England: an epi-demiological study. Br J Haematol 2013;161:525–32.
- Mor A, Shinar Y, Zaks N, et al. Evaluation of disease severity in familialMediterranean fever. Semin Arthritis Rheum 2005;35(1):57–64.
- Rezk T, Lachmann HJ, Fontana M, et al. Prolonged renal survival in light chainamyloidosis: speed and magnitude of light chain reduction is the crucial factor.Kidney Int 2017;92(6):1476-83.
- Рамеев В.В., Козловская Л.В., Рамеева А.С. и др. Особенности эволюции ипрогностическое значение поражения сердца у больных системным AL-амилоидозом. Клин фармакол тер 2019;29(2):49-57 [Rameev VV, KozlovskayaLV, Rameeva AS, et al. Evolution and prognostic value of heart disease in patientswith systemic AL-amyloidosis. Klinicheskaya farmakologiya i terapiya = ClinPharmacol Ther 2019;29(3):26-33 (In Russ.)].
- Cacoub P, Axler O, De Zuttere D, et al. Amyloidosis and cardiac involvement.Ann Med Interne (Paris) 2000;151: 611-617.
- Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment,diagnosis, and referral. Heart 2011;97(1):75-84.
- Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treat-ment in immunoglobulin light chain amyloidosis based on free light chain mea-surement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol2012;30:4541–9.
- Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system forlight chain amyloidosis incorporating cardiac biomarkers and serum free lightchain measurements. J Clin Oncol 2012;30:989–95.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiactransthyretin amyloidosis. Circulation 2016;133(24):2404-12.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003;349:583-96.
- Gertz MA, Kyle RA, Greipp PR. Response rates and survival in primary systemicamyloidosis. Blood 1991;77:257-62.
- Merlini G. CyBorD: stellar response rates in AL amyloidosis. Blood 2012;119:4343-5.
- Benditt EP, Eriksen N. Amyloid protein SAA is associated with high densitylipoprotein from human serum. Proc Natl Acad Sci USA 1977;74:4025–8.
- Cantarini L, Rigante D, Brizi MG, et al. Clinical and biochemical landmarks insystemic autoinflammatory diseases. Ann Med 2012;44(7):664–73.
- Hawkins PN, Lachmann HJ, Aganna E, McDermott MF. Spectrum of clinicalfeatures in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum2004;50(2):607–12.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med 1972;287(25):1302.
- Gattorno M, Sormani MP, D'Osualdo A, et al. A diagnostic score for molecularanalysis of hereditary autoinflammatory syndromes with periodic fever in children.Arthritis Rheum 2008;58(6):1823–32.
- Martinon F, Pétrilli V, Mayor A, et al. Gout-associated uric acid crystals activatethe NALP3 inflammasome. Nature 2006;440(7081):237–41.
- So A, De Smedt T, Revaz S, Tschopp J.A pilot study of IL-1 inhibition byanakinra in acute gout. Arthritis Res Ther 2007;9(2):R28.
- McGonagle D, Tan AL, Shankaranarayana S, et al. Management of treatmentresistant inflammation of acute on chronic tophaceous gout with anakinra. AnnRheum Dis 2007;66(12):1683–1684.
- Burger D, Dayer JM, Palmer G, Gabay C. Is IL-1 a good therapeutic target in thetreatment of arthritis? Best Pract Res Clin Rheumatol 2006;20(5):879–96.
- Tunca M, Kirkali G, Soytürk M, et al. Acute phase response and evolution offamilial Mediterranean fever. Lancet 1999;353(9162):1415.
- Mansfield E, Chae JJ, Komarow HD, et al. The familial Mediterranean fever pro-tein, pyrin, associates with microtubules and colocalizes with actin filaments.Blood 2001;98(3):851–59.
- Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety ofanakinra therapy in pediatric and adult patients with the autoinflammatoryMuckle-Wells syndrome. Arthritis Rheum 2011;63(3):840–9.
- Comenzo RL, Vosburgh E, Simms RW, et al. Dose-intensive melphalan withblood stem cell support for the treatment of AL amyloidosis: one-year follow-upin five patients. Blood 1996;88:2801-6.
- Dispenzieri A, Kyle RA, Lacy MQ, et al. Superior survival in primary systemicamyloidosis patients undergoing peripheral blood stem cell transplantation: a case-control study. Blood 2004;103(10):3960-3.
- Gundapanenia BK, Sultanb MB, Keohaneb DJ, Schwartz JH. Tafamidis delaysneurological progression comparably across Val30Met and non-Val30Met geno-types in transthyretin familial amyloid polyneuropathy. Eur J Neur 2018;25:464-8.
- Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patientswith transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379(11):1007-16.