Аутоиммунитет, аутовоспаление и почки
В статье рассматриваются основные варианты поражения почек при системных аутоиммунных и аутовоспалительных заболеваниях, в том числе гломерулонефрит, включая быстропрогрессирующий, тромботическая микроангиопатия, тубулоинтерстициальный нефрит и АА-амилоидоз. Частота и типы нефропатий при системных иммуновоспалительных заболеваниях существенно отличаются. Например, гломерулонефрит чаще всего встречается при системной красной волчанке и системных васкулитах с преимущественным поражением мелких сосудов, тромботическая микроангиопатия – при антифосфолипидном синдроме, в том числе вторичном, тубулоинтерстициальный нефрит – при синдроме Шегрена и IgG4-ассоциированной болезни, АА-амилоидоз – при ревматоидном артрите, анкилозирующем спондилите, псориатическом артрите, периодической болезни и некоторых других моногенных аутовоспалительных заболеваниях. Почечная выживаемость у пациентов со многими аутоиммунными и аутовоспалительными заболеваниями увеличилась благодаря более ранней диагностике и расширению арсенала иммуносупрессивных и противовоспалительных препаратов. Тем не менее, у части больных наблюдается прогрессирование хронической болезни почек, которое может быть и не связано с активностью основного заболевания.
Развитие системных воспалительных заболеваний отражает сложное взаимодействие аутоиммунных нарушений и аутовоспаления. Первые обусловлены активацией адаптивного (приобретенного) гуморального и/или клеточного иммунитета, в то время как аутовоспаление связано с генетически детерминированными нарушениями врожденного иммунитета [1,2]. Примерами преимущественно аутоиммунных заболеваний являются системная красная волчанка (СКВ), системные васкулиты, ассоциированные с антителами к цитоплазме нейтрофилов (АНЦА), антифосфолипидный синдром, ревматоидный артрит и др., в то время как аутовоспаление играет ключевую роль в развитии моногенных и некоторых полигенных аутовоспалительных заболеваний, таких как периодическая болезнь, криопирин-ассоциированный периодический синдром, болезнь Стилла взрослых и др. [3,4].
Поражение почек относится к числу ведущих проявлений системных аутоиммунных и аутовоспалительных заболеваний и обычно предполагает необходимость в усилении патогенетической терапии, учитывая высокую вероятность прогрессирующего ухудшения функции почек при сохранении активности болезни. В настоящее время результаты лечения системных воспалительных заболеваний значительно улучшились благодаря расширению арсенала противовоспалительных и иммуносупрессивных препаратов. Тем не менее, у части больных даже в случае достижения ремиссии основного заболевания отмечается продолжающееся снижение функции почек, которое может отражать естественную эволюцию нефропатии или воздействие других факторов, таких как артериальная гипертония или сахарный диабет. Развитие хронической почечной недостаточности, требующей лечения гемодиализом, сопровождается многократным увеличением риска сердечно-сосудистых осложнений, которые являются основной причиной смерти больных. Следует отметить и трудности дифференциальной диагностики поражения почек при системных воспалиантов, которые при сходстве лабораторных проявлений отличаются по течению и подходам к лечению.
Большой вклад в изучение поражения почек при системных аутоиммунных и аутовоспалительных заболеваниях внесли академики Е.М. Тареев и Н.А. Мухин, которые совместно с многочисленными учениками на протяжении нескольких десятилетий занимались разра боткой этой проблемы.
Варианты поражения почек при аутоиммунных и аутовоспалительных заболеваниях
Поражение почек наблюдается при любых системных аутоиммунных и многих аутовоспалительных заболева ниях, хотя частота его и клинические варианты суще ственно отличаются (табл. 1). При аутоиммунных заболеваниях развиваются все варианты диффузного поражения почек, включая гломерулонефрит, тубулоинтерстициальный нефрит или синдром каналь цевых нарушений, АА-амилоидоз и тромботическую микроангиопатию (ТМА), в то время как для аутовос палительных заболеваний характерно развитие АА-ами лоидоза. Исключением является подагра, однако при этом заболевании различные варианты поражения почек обусловлены в первую очередь гиперурикемией и гиперурикозурией, а не аутовоспалением как таковым. Необходимо учитывать, что поражение почек как при аутоиммунных, так и аутовоспалительных заболеваниях не всегда связано с основным патологическим процес сом и может быть следствием медикаментозной терапии, например, нестероидными противовоспали тельными препаратами (НПВП) или метотрексатом, или сопутствуюших заболеваний, таких как артериаль ная гипертония или сахарный диабет, в том числе вызванных глюкокортикостероидами.
| Аутоиммунные заболевания | Аутовоспалительные заболевания |
|---|---|
| Примечание: *в таблице указаны наиболее частые причины различных вариантов поражения почек. АФС - антифосфолипидный синдром, СКВ - системная красная волчанка, анти-БМК - антитела к базальной мембране клубочков, АНЦА - антитела к цитоплазме нейтрофилов. | |
Гломерулонефрит, в том числе быстропрогрессирующий
|
АА-амилоидоз
|
Среди аутоиммунных заболеваний поражение почек чаще всего встречается при системных васкулитах и СКВ. Причинами гломерулонефрита, включая быстро прогрессирующий, могут быть любые системные васку литы, поражающие преимущественно сосуды малого и среднего диаметра, в том числе АНЦА-ассоциирован ные васкулиты, криоглобулинемический васкулит, IgA васкулит и болезнь, ассоциированная с антителами к базальной мембране клубочков (анти-БМК болезнь, старое название – синдром Гудпасчера). При АНЦА ассоциированных васкулитах частота поражения почек составляет от 20 до 95% в зависимости от нозологиче ской формы. Среди 374 больных АНЦА-ассоцирован ными васкулитами, обследованных в клинике им. Е.М. Тареева, клинические признаки поражения почек чаще всего наблюдались у пациентов с микроскопическим полиангиитом (97%) и реже у пациентов с гранулемато зом с полиангиитом (44%) и эозинофильным грануле матозом с полиангиитом (23%). При гранулематозе с полиангиитом частота гломерулонефрита была ниже ожидаемой (60-80%), что отражало высокую долю боль ных с локальной формой заболевания в исследованной когорте [5]. Клинически АНЦА-ассоциированный гло мерулонефрит чаще всего проявлялся гематурией и протеинурией, которая сравнительно редко достигала нефротического уровня, а также нарастанием сыворо точного уровня креатинина. Более чем у трети больных наблюдалась картина быстропрогрессирующего гломе рулонефрита (БПГН). Более тяжелое течение АНЦА ассоциированного гломерулонефрита отмечалось у пациентов с микроскопическим полиангиитом, у кото рых были выше протеинурия, гематурия и исходный сывороточный уровень креатинина и часто развивалась быстропрогрессирующая почечная недостаточность (в 60% случаев). Хотя тяжелое поражение почек, в том числе БПГН, возможно у пациентов с гранулематозом с полиангиитом и эозинофильным гранулематозом с полиангиитом, тем не менее, в этих группах нередко наблюдался относительно благоприятный вариант гло мерулонефрита, проявлявшегося изолированным моче вым синдромом без значимого снижения функции почек или артериальной гипертонии [5,6]. Наличие антител к миелопероксидазе ассоциировалось с более тяжелым течения поражения почек, в то время у паци ентов с антителами к протеиназе 3 был выше риск раз вития обострений системного васкулита [7].
Поражение почек при IgA-васкулите (старые назва ния – пурпура Шенлейн-Геноха и геморрагический васкулит) часто встречается как у детей (20-54%), так и взрослых (45-85%) и характеризуется развитием микро гематурии (а также эпизодов макрогематурии), которая может сочетаться с протеинурией, чаще небольшой, и нарушением функции почек [8]. Развитие хронической почечной недостаточности у взрослых отмечается чаще, чем у детей. При криоглобулинемическом васкулите, который в большинстве случаев ассоциирован с HCV инфекцией, поражение почек развивается в 20-35% слу чаев [9] и проявляется микрогематурией и протеинурией субнефротического или нефротического уровня, которые нередко сопровождаются нарушением функции почек [10]. Как при IgA-васкулите, так и криоглобулинемическом васкулите возможно развитие БПГН, который наблюдается также у большинства пациентов с анти-БМК болезнью. Изменения в моче описаны и при артериите Такаясу, поражающем в основном крупные артерии, хотя они могут быть и не связаны с основным заболеванием [11]. В то же время при артериите Такаясу нередко поражаются почечные артерии, что приводит к развитию реноваскулярной гипертонии.
Поражение почек относится к основным висцераль ным проявлениям СКВ и встречается у 40% пациентов с этим заболеванием [12,13]. Еще чаще при СКВ определяются гистологические изменения в почках, которые могут не сопровождаться изменениями в моче. В нашей стране основные клинические варианты волчаночного нефрита были детально описаны И.Е. Тареевой в моно графии "Волчаночный нефрит", которая вышла в свет еще в 1976 г. Основные проявления волчаночного неф рита – протеинурия и микрогематурия. В отличие от поражения почек при системных васкулитах, при вол чаночном нефрите чаще наблюдается протеинурия суб нефротического уровня и нефротический синдром, которые выявляют примерно в половине случаев. Нередко встречаются канальцевые нарушения, которые могут быть следствием иммунокомплексного интерсти циального нефрита. У 2/3 больных наблюдается почеч ная недостаточность, которая может быть быстропрогрессирующей.
Возможные варианты поражения почек при ревматоидном артрите включают в себя гломерулонефрит, АА амилоидоз, ревматоидный васкулит и различные формы лекарственной нефропатии [14]. В отличие от СКВ, хронический гломерулонефрит при ревматоидном арт рите встречается редко, в то время как частота АА-ами лоидоза при этом заболевании, наоборот, выше [15]. В прошлом важную роль в этиологии поражения почек у больных ревматоидным артритом играли нефротоксич ные лекарственные средства, такие как D-пеницилла мин, циклоспорин и препараты золота, однако в последующем они были заменены более безопасными базисными противовоспалительными противовоспали тельными препаратами (БПВП) и генно-инженерными биологическими препаратами (ГИБП) [16]. В то же время многие пациенты с ревматоидным артритом по прежнему годами принимают нестероидные противо воспалительные препараты, которые могут вызвать интерстициальный нефрит.
У 10% больных системной склеродермией развивается почечный криз, характеризующийся внезапным появлением артериальной гипертонии, в том числе зло качественной, и почечной недостаточности при отсут ствии признаков гломерулонефрита [17]. Причиной почечного криза считают ухудшение перфузии почек вследствие вазоспазма и сужения почечных артериол, которые нередко осложняются тромбозом (ТМА).
| Заболевание | Клинические проявления | Характерные морфологические изменения в ткани почек |
|---|---|---|
| Примечание: *биопсия почки у пациентов с узелковым полиартериитом сопряжена с риском кровотечения ввиду возможного развития аневризм в почечных артериях. ГН – гломерулонефрит, ОПП – острое повреждение почек, СКФ – скорость клубочковой фильтрации | ||
| АНЦА-ассоциированные васкулиты | Острый или быстропрогрессирующий нефритический синдром |
Экстракапиллярный ГН III типа (с полулуниями, мало/олигоиммунный) |
| Анти-БМК болезнь | Острый или быстропрогрессирующий нефритический синдром |
Экстракапиллярный ГН I типа (с полулуниями, линейное отложение иммунных депозитов) |
| Криоглобулинемический васкулит | Мочевой синдром Нефротический синдром Острый нефритический синдром Их сочетание |
Мембранопролиферативный ГН I типа (иммунокомплексный) Экстракапиллярный ГН II типа (с полулуниями, гранулярное отложение иммунных депозитов) |
| IgA-васкулит | Мочевой синдром Редко: нефротический или острый нефритический синдром |
IgA-нефропатия (мезангиопролиферативный ГН с отложением IgA) Редко: Экстракапиллярный ГН II типа |
| Узелковый полиартериит | Прогрессирующее снижение СКФ, в том числе ОПП Протеинурия |
Ишемические изменения клубочков в отсутствие пролиферации и иммунных депозитов* |
| СКВ | Мочевой синдром Нефротический синдром Острый нефритический синдром |
Волчаночный нефрит 1-6 классов Тубулоинтерстициальный нефрит Волчаночная подоцитопатия Васкулит почечных сосудов |
| Системная склеродермия | ОПП (склеродермический почечный криз) Прогрессирующая ХБП |
Васкулопатия мелких артерий и/или артериол, ТМА |
| Синдром Шегрена | Синдром канальцевых нарушений (электролитные нарушения, канальцевая протеинурия,почечный канальцевый ацидоз) Снижение СКФ Редко: острый нефритический синдром |
Тубулоинтерстициальный нефрит Редко: мембранопролиферативный ГН I типа |
| Антифосфолипидный синдром | Снижение СКФ, в том числе с развитием ОПП Протеинурия |
ТМА |
| IgG4-ассоциированное заболевание | Синдром канальцевых нарушений Снижение СКФ, в том числе с развитием ОПП Протеинурия, нефротический синдром |
Тубулоинтерстициальный нефрит Мембранозная нефропатия |
| Воспалительные заболевания суставов (ревматоидный артрит, псориатический артрит, анкилозирующий спондилит) |
Протеинурия с последующим формированием нефротического синдрома Снижение СКФ |
АА-амилоидоз Редко: пролиферативные формы ГН |
| Подагра | Нефролитиаз Синдром канальцевой дисфункции Снижение СКФ |
Тубулоинтерстициальный нефрит |
| Периодическая болезнь, TRAPS, криопирин-ассоциированный периодический синдром, гипериммуноглобулинемия D |
Протеинурия, нефротический синдром, снижение СКФ |
АА-амилоидоз |
Наиболее частые проявления поражения почек при системных аутоиммунных и аутовоспалительных заболеваниях приведены в (табл. 2).
Быстропрогрессирующий гломерулонефрит
Быстропрогрессирующий гломерулонефрит (БПГН) – это наиболее тяжелый вариант гломерулонефрита, требующий неотложного лечения [18]. Характеризуется наличием остронефритического синдрома и быстрым прогрессированием почечной недостаточности (удвое ние сывороточного содержания креатинина в два раза за 3 мес). Морфологическим субстратом БПГН обычно является экстракапиллярный гломерулонефрит с кле точными или фиброзно-клеточными полулуниями, которые определяются по крайней мере в 50% клубоч ков (некоторые авторы считают возможным диагности ровать это состояние при наличии полулуний в 10-50% клубочков). Полулуния образуются в результате повреждения клубочков и разрыва стенок капилляров, который приводит к проникновению плазменных бел ков и воспалительных клеток в пространство капсулы Шумлянского-Боумена. Процесс повреждения клубоч ков может инициироваться АНЦА, антителами к БМК или циркулирующими иммунными комплексами. Разрыв капсулы вызывает поступление из интерстиция фибробластов и миофибробластов, которые синтези руют матриксные белки (коллагены I и III типов, фиб ронектин), что ведет к фиброзу полулуний и необратимой утрате функции почек. Важное значение имеет своевременная диагностика БПГН, так как агрес сивная иммуносупрессивная терапия глюкортикосте роидами и циклофосфамидом или ритуксимабом, а также плазмаферез позволяют остановить прогрессирование почечной недостаточности, а в части случаев добиться полного восстановления функции почек.
Важное значение в диагностике БПГН и установлении его причины имеет серологическое исследование, в том числе определение АНЦА и анти-БМК, а также антинуклеарного фактора и криоглобулинов. Необходимо учитывать, что у 5-9% пациентов с АНЦА-ассо циированным васкулитом определяются анти-БМК, в то время как АНЦА, прежде всего к миелопероксидазе, выявляют примерно у трети пациентов с анти-БМК болезнью [21]. Клинический фенотип у пациентов с двумя типами антител сходен с таковым анти-БМК болезни без АНЦА, включая частое развития тяжелого поражения почек в начале заболевания, хотя у них могут наблюдаться и некоторые черты, свойственные АНЦА-ассоциированному васкулиту, такие как пожилой возраст, наличие внепочечных проявлений помимо поражения легких, более высокая вероятность восста новления функции почек и риск рецидивов, которые обычно отсутствуют при анти-БМК болезни.
Для подтверждения диагноза БПГН всем больным рекомендуется проводить биопсию почки. В зависимо сти от гистологической картины выделяют три типа экстракапиллярного гломерулонефрита с полулуниями: I тип (антительный) – линейное свечение антител в почечном биоптате и наличие анти-БМК в крови; II тип (иммунокомплексный) – гранулярный тип свече ния в биоптате при отсутствии циркулирующих АНЦА и анти-БМК; III тип (малоиммунный) – свечение иммуноглобулинов и комплемента в биоптате отсутствует или незначительно выражено. I тип наблюдается при анти-БМК болезни, II тип характерен для БПГН, связанного с инфекциями (постстрептококковый БПГН), СКВ, криоглобулинемией, IgA-васкулитом, III тип развивается при АНЦА-ассоциированных васкули тах [18]. По данным крупного американского исследо вания, в которое были включены 632 пациента с гистологически подтвержденным экстракапиллярным гломерулонефритом с полулуниями, в 60% случаев определялся малоиммунный гломерулонефрит (III типа), в 24% – иммунокомплексный (II типа) и в 15% – антительный (I типа) [19]. В турецком исследовании АНЦА-ассоциированные васкулиты (преимущественно микроскопический полиангиит и гранулематоз с поли ангиитом) были диагностированы у 56,5% из 200 боль ных БПГН, подтвержденным при биопсии почки, IgA-нефропатия – у 7,0%, анти-БМК болезнь – у 5,5%, волчаночный нефрит – у 1,5%, постстрептококковый БПГН – у 1,5% [22].
Тромботическая микроангиопатия
Тромботическая микроангиопатия (ТМА) – это клинико-морфологический синдром, характеризующийся повреждением эндотелия, накоплением аморфного материала в субэндотелиальном пространстве и образо ванием тромбов в сосудах микроциркуляторного русла, что приводит к их окклюзии и ишемии органов и тка ней, прежде всего почек [23]. Типичные клинические проявления ТМА включают в себя тромбоцитопению (<150 × 109/л или снижение количества тромбоцитов по крайней мере на 25% от исходного), микроангиопати ческую гемолитическую анемию (МАГА) и острое повреждение почек (ОПП), которое в 2/3 случаев при водит к развитию хронической почечной недостаточно сти, требующей заместительной терапии [24]. Причиной МАГА являются механическое повреждение и разрушение эритроцитов при контакте их с тромбами в сосудах микроциркуляторного русла. Помимо Кумбс негативной анемии, к признакам внутрисосудистого гемолиза относят увеличение количества фрагментиро ванных эритроцитов (шистоцитов) более >0,1%, повышение активности ЛДГ и снижение содержания гаптоглобина, связывающего гемоглобин.Первичная ТМА включает в себя тромботическую тромбоцитопе ническую пурпуру, типичный гемолитико-уремический синдром (STEC-ГУС) и атипичный ГУС, в то время как вторичная ТМА развивается под действием различных факторов, в том числе лекарств, инфекций, опухолей, беременности, трансплантации органов [25].
Среди аутоиммунных заболеваний основной причиной ТМА является антифосфолипидный синдром (АФС) [26]. Наиболее тяжелый вариант этого состояния – катастрофический АФС, который характеризуется поражением не только почек, но и других органов с развитием полиорганной недостаточности. Необходимо учитывать, что при АФС встречаются и другие вариан ты поражения почек, в частности односторонний или двусторонний стеноз или тромбоз почечных артерий, сопровождающийся реноваскулярной артериальной гипертонией и ишемической нефропатией, тромбоз почечных вен, редко гломерулонефрит [27]. Течение ТМА может быть не только острым, но и хроническим. В последнем случае при биопсии почки выявляют река нализированные тромбы в артериолах, артериосклероз, гиперплазию интимы почечных артериол и их фибро пластическую окклюзию, атрофию канальцев, в то время как характерным гистологическим признаком острой ТМА является наличие микротромбов в почеч ных артериолах и/или клубочках при отсутствии признаков воспаления и иммунных депозитов при иммунофлюоресцентом исследовании [27].
ТМА наблюдается как при первичном АФС, так и вторичном АФС у пациентов с СКВ. V. Domingues и соавт. провели мета-анализ 36 исследований у 3035 пациентов с СКВ, у 454 из которых имелись признаки острой или хронической ТМА [28]. Частота поражения микрососудов почек у больных, у которых определялись и не определялись антифосфолипидные антитела, составила 31,3% и 10,4%, соответственно. Наличие антифосфолипидных антител сопровождалось увеличе нием риска развития ТМА в 3,0 раза, в том числе острой – в 2,9 раза и хронической – в 2,6 раза. Риск острого или хронического повреждения микрососудов почек был достоверно повышен при наличии волчаноч ного антикоагулянта и IgG антител к кардиолипину, в то время как при наличии антител к b2-гликопротеину данная ассоциация не достигла статистической значи мости. У пациентов с СКВ возможно сочетание волча ночного нефрита и ТМА, что подчеркивает важность биопсии почек для установления варианта поражения почек при этом заболевании [29].
Признаки ТМА нередко наблюдаются у пациентов со склеродермическим почечным кризом, в основе патогенеза которого лежат эндотелиальная дисфункция и повреждение почечных сосудов, вызывающие активацию коагуляции и образование тромбов в микрососудах [17]. ТМА встречается и при других аутоиммунных заболеваниях, в том числе АНЦА-ассоциированном васкулите. Во французский регистр включены 41 паци ент с ТМА, ассоциированной с СКВ (n=18), синдромом Шегрена (n=7), системной склеродермией (n=11), сме шанными заболеваниями соединительной ткани (n=2) и системными васкулитами (n=2) [30].
АА-амилоидоз
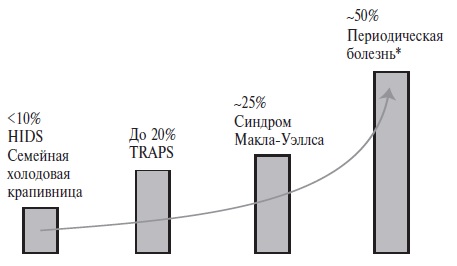
Поражение почек наблюдается при различных типа амилоидоза, прежде всего AL, AA и ATTR (транстире тиновом). Предшественником АА-амилоида является сывороточный амилоидный белок (SAA), который представляет собой белок острой фазы, сходный с С-реактивным белком [31]. Концентрации двух белков коррелируют друг с другом и увеличиваются при воспа лении в результате усиления синтеза в гепатоцитах под действием интерлейкина (ИЛ)-6 и ИЛ-1β. Соответ ственно, АА-амилоидоз может развиться при системных аутоиммунных и аутовоспалительных заболеваниях, особенно при сохранении высокой активности, кото рую не удается снизить противовоспалительными и/или иммуносупрессивными препаратами. Хотя случаи АА амилоидоза описаны при любых аутоиммунных заболе ваниях, тем не менее, для многих из них развитие его не характерно. Например, АА-амилоидоз очень редко встречается у пациентов с СКВ или АНЦА-ассоцииро ванными и другими васкулитами [32,33], хотя мы наблюдали несколько случаев вторичного амилоидоза в когорте из 128 больных артериитом Такаясу [11]. Среди аутоиммунных заболеваний основной причиной АА амилоидоза остается ревматоидный артрит. В наших исследованиях доля его в структуре этиологических факторов АА-амилоидоза составляла около трети [33], а в этиологии нефропатии у больных ревматоидным арт ритом – примерно 40% [15]. Нередкими причинами АА-амилоидоза сегодня являются полигенные аутовос палительные заболевания, такие как анкилозирующий спондилит, псориатический артрит, болезнь Стилла взрослых, подагрический артрит, а также моногенные аутовоспалительные заболевания, в том числе периодическая болезнь (семейная средиземноморская лихорадка), криопирин-ассоциированный периодический синдром, гипериммуноглобулинемия D и TRAPS (рис. 1) [34,35]. В нашем исследовании основным предикто ром развития АА-амилоидоза у 170 пациентов с перио дической болезнью был рецидивирующий экссудативный артрит (отношение шансов 2,28; 95% доверительный интервал 1,17–4,42) [36]. Обращает на себя внимание, что поражение суставов характерно и для других заболеваний, которые сегодня занимают ведущее место в этиологии АА-амилоидоза, что может отражать продукцию предшественника амилоида синовиальной оболочкой. R. O’Hara и соавт. выявили высокую экспрессию мРНК SAA синовиальными клетками и повышенную концентрацию SAA в синовиальной жидкости у пациентов с активным ревматоидным артритом [37].
Эпидемиология амилоидоза за последние десятилетия существенно изменилась. По данным Британского национального центра по изучению амилоидоза, общее количество случаев этого заболевания на протяжении последних 10 лет увеличилось в несколько раз по сравнению с таковым в 1987-1999 гг. [38]. На протяже нии всего указанного периода доля AL-амилоидоза в структуре зарегистрированных случаев оставалась ста бильной (более 50%), в то время как доля AA-амилои доза значительно снизилась и в 2016-2019 гг. составляла всего 3%. В ряде исследование показано снижение частоты АА-амилоидоза при ревматоидном артрите и других системных воспалительных заболеваниях, в том числе спондилоартрите и периодической болезни [39,40]. Указанные изменения отражают успехи в лечении хронических воспалительных заболеваний, связанные с применением БПВП и ГИБП [41].
Тубулоинтерстициальный нефрит
Системные аутоиммунные заболевания могут приводить к повреждению не только структур клубочка, но и почечных канальцев, вследствие развития тубулоинтер стициального нефрита (ТИН) или ишемии при вовлечении в патологический процесс мелких сосудов (например, в рамках АФС). Вклад поражения тубулоин терстиция в развитие почечной дисфункции нередко недооценивают в клинической практике у пациентов с АНЦА-ассоциированными васкулитами, СКВ и други ми заболеваниями, сопровождающимися развитием гломерулонефрита. Однако формирование атрофии канальцев и интерстициального фиброза вследствие активного ТИН является значимым предиктором неблагоприятного почечного исхода при гломеруляр ных заболеваниях [42]. В связи с этим оценка выражен ности воспаления и склероза интерстиция в биоптате проводится при подсчете значений индексов активно сти и хронизации у пациентов с волчаночным нефри том [43], при анализе риска развития терминальной почечной недостаточности у пациентов с АНЦА-ассо циированными васкулитами (ANCA renal risk score) [44] и IgA-нефропатией [45].
При ряде заболеваний, в частности при синдроме Шегрена, IgG4-ассоциированном заболевании, саркои дозе и подагре, ТИН может быть ведущим проявлением поражения почек, а тяжесть его варьирует от умеренных проявлений синдрома канальцевой дисфункции до ОПП. Широкое разнообразие клинических проявлений поражения тубулоинтерстиция можно наблюдать у пациентов с синдромом Шегрена [46]. В числе первых проявлений канальцевой дисфункции при синдроме Шегрена развиваются электролитные нарушения, иногда с формированием развернутой картины дистального почечного канальцевого ацидоза (гипокалиемия, мета болический ацидоз с положительным анионным промежутком в моче). Также характерны нарушения концентрационной функции почек (никтурия, полиу рия, стойкое снижение относительной плотности мочи, редко – развитие нефрогенного несахарного диабета). В редких случаях у пациентов с синдромом Шегрена возможно развитие проксимального канальцевого ацидоза, приобретенных синдромов Барттера и Гительмана. У части пациентов высокая активность ТИН может приводить к прогрессирующему снижению скорости клубочковой фильтрации (СКФ), иногда с развитием ОПП.
Поскольку многие клинические проявления ТИН неспецифичны (электролитные нарушения, умеренная протеинурия, снижение СКФ), основным методом подтверждения диагноза является биопсия почки. Помимо оценки активности воспаления и выраженности тубу лоинтерстициального фиброза морфологическое иссле дование ткани почки позволяет обнаружить патогномоничные для некоторых заболеваний особен ности, в частности специфические паттерны фиброза и инфильтрацию IgG4-позитивными плазматическими клетками при IgG4-ассоциированном заболевании [47], отложение кристаллов моноурата натрия и умеренную выраженность воспалительной инфильтрации при уратной нефропатии [48], специфические гранулемы при саркоидозе [49], а сопутствующие признаки поражения сосудов и ТМА в биоптате могут указывать на наличие АФС [50].
Роль биопсии в диагностике поражения почек приаутоиммунных и аутовоспалительных заболеваниях
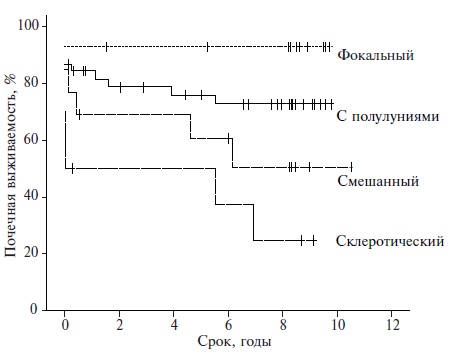
Аутоиммунные заболевания могут приводить к поражению любых структур ткани почки: клубочков, канальцев и сосудов различного калибра, а при ряде заболеваний (СКВ, системные васкулиты) нередко – к их сочетанному вовлечению. Этим объясняется широкий спектр клинических проявлений поражения почек, которое может даже при одном заболевании (например, СКВ) варьировать от бессимптомного течения или изолированной гематурии до тяжелого нефротического синдрома и фульминантного развития почечной недостаточности. При этом клинические проявления почечного процесса при многих заболеваниях могут не в полной мере отражать тяжесть гистологических изменений в ткани почки, что создает предпосылки для диагностических ошибок и выбора неоптимальной тактики ведения в клинической практике [51]. Биопсия почки остается "золотым стандартом" диагностики и оценки прогноза поражения почек, а ее выполнение показано существенной доле пациентов с аутоиммунными заболеваниями при появлении признаков поражения почек. В качестве примера рис. 2 демонстрирует зависимость почечной выживаемости от морфологического варианта АНЦА-ассоциированного гломерулонефрита [52].
Выполнение биопсии почки при ряде заболеваний регламентировано существующими клиническими рекомендациями. В частности, у пациентов с СКВ рекомендации EULAR/ERA-EDTA предусматривают проведение биопсии почки при наличии гематурии и/или зернистых цилиндров в моче, протеинурии >500 мг/сут, снижения СКФ [12], а рекомендации KDIGO – при протеинурии ≥500 мг/сут в сочетании с изменениями мочевого осадка (или без них), а также при снижении СКФ, которое нельзя объяснить иными причинами [53].
При отсутствии четких указаний на необходимость выполнения биопсии почки в клинических рекомендациях при том или ином заболевании следует ориентироваться на универсальные показания к выполнению морфологического исследования ткани почки:- Острый нефритический синдром (гематурия, протеинурия <3,5 г/сут, повышение уровня креатинина в сыворотке и снижение СКФ, отеки, артериальная гипертония).
- Нефротический синдром (протеинурия >3,5 г/сут, гипоальбуминемия, гипопротеинемия, гиперхолестеринемия, отеки).
- ОПП неясной этиологии, которое не разрешается в течение 7-14 дней.
- Отторжение и дисфункция трансплантата почки.
- Протеинурия >0,5 г/сут.
- Клубочковая гематурия.
- Прогрессирование ХБП, этиология которой не ясна (снижение СКФ на ≥5 мл/мин за год).
Лечение и профилактика поражения почек при аутоиммунных и аутовоспалительных заболеваниях
За последние десятилетия достигнуты значительные успехи в лечении аутоиммунных и аутовоспалительных заболеваний, что привело к снижению риска развития терминальной хронической почечной недостаточности у пациентов с различными вариантами поражения почек. Например, в когортном исследовании, в котором изучались почечные исходы у 499 пациентов с волчаночным нефритом на протяжении 50 лет, было выявлено увеличение 10-летней почечной выживаемости с 87% в 1970-1985 гг. до 99% в 2002-2016 гг. [54]. Кроме того, авторы отметили снижение частоты нарушения функции почек в начале волчаночного нефрита, более позднее его развитие и увеличение частоты изолированных изменений в моче. R. Rhee и соавт. изучали почечные исходы и смертность у 554 пациентов с АНЦА-ассоциированными васкулитами с поражением почек, диагностированными с 1985 по 2009 г. [55]. Общая 5-летняя выживаемость увеличилась с 72% в 1985-1989 гг. до 83% в 2005-2009 гг., а 5-летняя почечная выживаемость – с 65% до 82%. Единственным независимым предиктором развития терминальной хронической почечной недостаточности или смерти было исходное сывороточное содержание креатинина, что указывало на важность ранней диагностики системного васкулита для улучшения его исходов. В недавно опубликованном исследовании у 123 пациентов с антиБМК болезнью 5-летняя почечная выживаемость составила всего 34%, однако у пациентов, у которых диагноз был установлен после 2007 г., она была в два раза выше, чем у больных, у которых заболевание было выявлено до указанного срока [56]. Факторами, позволявшими предсказать очень низкую вероятность восстановления функции почек, были необходимость в лечении диализом в дебюте заболевания, наличие клеточных полулуний во всех клубочках и склероза более чем в половине клубочков. Улучшение почечной выживаемости у пациентов с системными аутоиммунными заболеваниями отражает внедрение в клиническую практику современных схем патогенетической терапии, которые предполагают применение не только глюкокортикостероидов, но и иммуносупрессивных препаратов (циклофосфамида, микофенолата мофетила, азатиоприна и др.) и/или ГИБП (прежде всего ритуксимаба), а также плазмообмена у отдельных категорий пациентов [13,57,58]. Немаловажное значение имеет и более ранняя диагностика аутоиммунных заболеваний. В настоящее время продолжается разработка новых препаратов для лечения иммуновоспалительных ревматических заболеваний. Например, недавно было одобрено применение воклоспорина (ингибитора кальциневрина) и внутривенного белимумаба (анти-В-клеточного ГИБП) для лечения волчаночного нефрита [59]. Перспективными препаратами при этом заболевании считают также обинутузумаб (анти-CD20 антитела), даратумумаб (анти-CD38 антитела), сиролимус (ингибитор mTOR) и др. В опытах на мышах авакопан, блокирующий рецепторы С5а компонента комплемента, предупреждал развитие гломерулонефрита, вызванного антителами к миелопероксидазе [60], а в клиническом исследовании ADVOCATE этот препарат оказывал благоприятное влияние на альбуминурию и расчетную СКФ у пациентов с АНЦАассоциированными васкулитами [61]. Ингибиторы комплемента изучаются и у пациентов с СКВ, в том числе волчаночным нефритом [62].
Препаратов для лечения АА-амилоидоза в настоящее время не существует, однако вполне реальной задачей при аутоиммунных и аутовоспалительных заболеваниях является профилактика его развития и прогрессирования путем эффективного подавления воспаления и, соответственно, продукции предшественника АА-амилоида. Необходимо учитывать, что образование амилоида может продолжаться при сохранении даже субклинического воспаления. У больных РА развитие АА-амилоидоза ассоциировалось с большей длительностью заболевания (>15 лет) и недостаточным контролем воспаления, характеризовавшимся стойким повышением уровня СРБ (>15 мг/л) [63]. В крупном турецком исследовании персистирование воспалительной активности (увеличение содержания СРБ) в межприступный период, которое отмечалось у 15% из 917 больных периодической болезнью несмотря на лечение колхицином, сопровождалось увеличением риска развития амилоидоза в 3,6 раза (p<0,001) [64]. Адекватная патогенетическая терапия имеет особое значение при наличии признаков амилоидоза (нарастающая протеинурия, нефротический синдром), учитывая вероятность его прогрессирования и развития хронической почечной недостаточности, требующей заместительной почечной терапии. У пациентов с ревматоидным артритом, осложнившимся АА-амилоидозом, перспективно применение ингибиторов интерлейкина-6, которые вызывают быстрое снижение содержания острофазных белков, в том числе SAA, и в части случаев позволяют стабилизировать или даже улучшить функцию почек [65]. Влияние ГИБП на течение АА-амилоидоза у пациентов с анкилозирующим спондилитом мало изучено, хотя опубликован положительный опыт применения ингибиторов фактора некроза опухоли a у таких больных в небольшом неконтролируемом исследовании [66]. У пациентов с моногенными аутовоспалительными заболеваниями, осложнившимися АА-амилоидозом, эффективное лечение, начатое до развития терминальной хронической почечной недостаточности, в большинстве случаев привело к регрессу амилоидоза или стабилизации функции почек [67].
Основным медиатором аутовоспаления, прежде всего при инфламасоммопатиях, является ИЛ-1β, что послужило основанием для изучения эффективности ингибиторов этого цитокина, в частности канакинумаба (моноклонального антитела, взаимодействующего с ИЛ-1β), при различных аутовоспалительных заболеваниях. Эффективность и безопасность канакинумаба подтверждены в двойных слепых, плацебо-контролируемых исследованиях, которые проводились у больных колхицинрезистентной периодической болезнью, гипериммуноглобулинемией D/недостаточностью мевалонаткиназы и TRAPS (исследование CLUSTER), криопирин-ассоциированным периодическим синдромом и болезнью Стилла взрослых (исследование CONSIDER) [41]. В этих исследованиях лечение канакинумабом вызывало не только купирование основных проявлений аутовоспалительных заболеваний, но и снижение содержания SAA, что имеет важное значение для профилактики АА-амилоидоза (рис. 3).
![Изменение доли (%) пациентов с различными уровнями SAA при лечении канакинумабом у больных колхицин-резистентной периодической болезнью, гипериммуноглобулинемией D/недостаточностью мевалонаткиназы и TRAPS в исследовании CLUSTER [73]](/wp-content/uploads/2022/12/autoimmunitet-autovospalenie-i-pochki_fig3.jpg)
Несмотря на лечение у части больных нефропатией любого происхождения развивается терминальная хроническая почечная недостаточность. По данным метаанализа 187 исследований более чем у 18000 пациентов с волчаночным нефритом, доля больных, которым через 5 лет потребовалась заместительная почечная терапия, составила 11%, а среди пациентов с IV классом волчаночного нефрита – 19% [68]. Через 10 лет доля таких больных достигла 17% и 33%, соответственно. В когортном исследовании у 523 пациентов с АНЦАассоциированными васкулитами частота развития терминальной хронической почечной недостаточности в течение 3 лет достигла 26% [69]. Обращает на себя внимание, что у 43% пациентов прогрессирование ХБП отмечалось несмотря на отсутствие признаков активности системного васкулита. Приведенные данные свидетельствуют о том, что прогрессирование ХБП у пациентов с системными аутоиммунными заболеваниями далеко не всегда связано с тяжелым течением гломерулонефрита и/или неадекватностью иммуносупрессивной терапии (позднее начало, неадекватный выбор препаратов или их доз и т.п.) и может отражать естественную эволюцию заболевания, характеризующуюся постепенным нарастанием склеротических изменений в почках. В связи с этим важное значение для профилактики ухудшения функции почек имеют контроль факторов, способствующих прогрессированию ХБП (артериальная гипертония, гипергликемия, гиперурикемия, избыточная масса тела и др.) и применение нефропротективных препаратов [70]. С целью нефропротекции чаще всего применяют блокаторы ренин-ангиотензин-альдостероновой системы, эффективность которых была установлена более 20 лет назад. Новыми препаратами, обладающими нефропротективной активностью, являются ингибиторы натрийглюкозного транспортера 2 типа (НГЛТ-2). В рандомизированном плацебо-контролируемом исследовании DAPA-CKD, в которое были включены 4304 пациента с ХБП (расчетная СКФ 25-75 мл/мин/1,73 м2 и отношение альбумин/креатинин в моче 200-5000 мг/г), лечение дапаглифлозином в дозе 10 мг/сут в течение среднем 2,4 лет привело к снижению риска комбинированной конечной точки, включавшей в себя стойкое снижение СКФ по крайней мере на 50%, развитие терминальной хронической почечной недостаточности или смерть от почечных или сердечно-сосудистых причин, на 39% (отношение рисков 0,61; 95% доверительный интервал 0,51-0,72) по сравнению с плацебо [71]. Следует отметить, что применение ингибитора НГЛТ-2 в этом исследовании вызывало снижение риска не только почечных, но и сердечно-сосудистых исходов, которые остаются основной причиной смерти пациентов с ХБП. Из исследования исключали пациентов с волчаночным нефритом или АНЦА-ассоциированным васкулитом. Тем не менее, применение ингибиторов НГЛТ-2 у части пациентов с ХБП, развившейся в исходе указанных заболеваний, представляется оправданным, особенно при наличии дополнительных показаний к их назначению, таких как сахарный диабет 2 типа [72].
Заключение
Частота и характер поражения почек при системных аутоиммунных и аутовоспалительных заболеваниях варьируются в широких пределах. Клинические и лабораторные проявления нефропатий различного происхождения (гломерулонефрит, в том числе быстропрогрессирующий, острая и хроническая ТМА, канальцевые нарушения, АА-амилоидоз почек) сходные, поэтому для их дифференциальной диагностики обычно необходимо гистологическое исследование биоптата почки, позволяющее выбрать адекватное патогенетическое лечение. Важное значение имеет ранняя диагностика поражения почек при аутоиммунных и аутовоспалительных заболевания, так как даже адекватная иммуносупрессивная и/или противовоспалительная терапия, но начатая при наличии необратимых склеротических изменений в почечных клубочках и канальцах, не позволяет предотвратить прогрессирование ХБП.
Используемые источники
- Szekanecz Z, McInnes IB, Schett G, et al. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat Rev Rheumatol 2021;17:585-95.
- Рамеев В.В., Лысенко (Козловская) Л.В., Богданова М.В., Моисеев С.В. Аутовоспалительные заболевания. Клин фармакол тер 2020;29(4):49-60 [Rameev V, Lysenko (Kozlovskaya) L, Bogdanova M, Moiseev S. Autoinflammatory diseases. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2020;29(4):49-60 (In Russ.)].
- Betrains A, Staels F, Schrijvers R, et al. Systemic autoinflammatory disease in adults. Autoimmun Rev 2021;20:102774.
- Моисеев С.В., Рамеев В.В. Дифференциальный диагноз системных аутовоспалительных заболеваний. Клин фармакол тер 2022;31(2):5-13 [Moiseev S, Rameev V. Differential diagnosis of systemic autoinflammatory diseases. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(2):5-13 (In Russ.)].
- Буланов Н.М., Моисеев С.В., Новиков П.И. и др. Поражение почек при различных вариантах АНЦА-ассоциированных васкулитов. Клин фармакол тер 2016;25(5):23-9 [Bulanov NM, Moiseev SV, Novikov PI, et al. Renal involvement in ANCA-associated vasculitis. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2016;25(5):23-9 (In Russ.)].
- Загвоздкина Е.С., Новиков П.И., Моисеев С.В. Особенности клинических проявлений и течения эозинофильного гранулематоза с полиангиитом в зависимости от наличия антител к цитоплазме нейтрофилов. Клин фармакол тер 2017;26(1):24-30 [Zagvozdkina E, Novikov P, Moiseev S. Clinical features of eosinophilic granulomatosis with polyangiitis in ANCA-positive and ANCA-negative patients. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2017;26(1):24-30 (In Russ.)].
- Буланов Н.М., Макаров Е.А., Щеголева Е.М. и др. Взаимосвязь антительного профиля и клинического течения поражения почек при АНЦА-ассоциированных васкулитах. Терапевтический архив 2018;90(6):16-21 [Bulanov N, Makarov E, Shchegoleva E, et al. Relationship between serologic profile (ANCA type) and clinical features of renal involvement in ANCA-associated vasculitides. Terapevticheskii Arkhiv 2018;90(6):16-21 (In Russ.)].
- Pillebout E, SunderkЪtter C. IgA vasculitis. Semin Immunopathol 2021;43(5):729-738.
- Cacoub P, Comarmond C, Domont F, et al. Cryoglobulinemia vasculitis. Am J Med 2015;128(9):950-5.
- Roccatello D, Fornasieri A, Giachino O, et al. Multicenter study on hepatitis C virus-related cryoglobulinemic glomerulonephritis. Am J Kidney Dis 2007;49(1):69-82.
- Мухин Н.А., Смитиенко И.О., Новиков П.И. и др. Артериит Такаясу: трудности диагностики, лечение и исходы в когортном исследовании у 128 больных. Клин фармакол тер 2014;23(3):55-61 [Mukhin NA, Smitienko IO, Novikov PI, et al.Clinical manifestations and outcomes of Takayasu arteritis in the cohort study in 128 patients. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2014;23(3):55-61 (In Russ.)].
- Fanouriakis A, Kostopoulou M, Cheema K, et al. 2019 Update of the Joint European League Against Rheumatism and European Renal AssociationEuropean Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendationsfor the management of lupus nephritis. Ann Rheum Dis 2020;79(6): 713-23.
- Бобкова И.Н., Моисеев С.В., Лысенко Л.В., Камышова Е.С. Волчаночный нефрит в XXI веке. Терапевтический архив 2022;94(6):713–7 [Bobkova IN, Moiseev SV, Lysenko LV, Kamyshova ES. Lupus nephritis in the XXI century. Terapevticheskii Arkhiv 2022;94(6):713–7 (In Russ.)].
- Kapoor T, Bathon J. Renal manifestations of rheumatoid arthritis. Rheum Dis Clin North Am 2018;44(4):571-84.
- Чеботарева Н.В., Гуляев С.В., Андросова Т.В. и др. Клинико-морфологические варианты и факторы риска поражения почек при ревматоидном артрите. Терапевтический архив 2020;92(5):55–60 [Chebotareva NV, Gulyaev SV, Androsova TV, et al. Clinicopatological variants and risk factors for chronic kidney disease in rheumatoid arthritis. Terapevticheskii Arkhiv 2020;92(5):55–60 (In Russ.)].
- Моисеев С.В., Новиков П.И., Чеботарева Н.В. и др. Внесуставные (системные) проявления ревматоидного артрита. Клин фармакол тер 2020;29(1):53-60 [Moiseev SV, Novikov PI, Chebotareva NV, et al. Extraarticular (systemic) manifestations of rheumatoid arthritis. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2020;29(1):53-60 (In Russ.)].
- Reggiani F, Moroni G, Ponticelli C. Kidney involvement in systemic sclerosis. J Pers Med 2022;12(7):1123.
- Шилов Е.М., Козловская Н.Л., Коротчаева Ю.В. Клинические рекомендации по диагностике и лечению быстропрогрессирующего гломерулонефрита (экстракапиллярного гломерулонефрита с полулуниями). Нефрология 2015;19(6):73–82 [Shilov EM, Kozlovskaya NL, Korotchaeva JV. Clinical guidelines for diagnosis and treatment of rapidly progressive glomerulonephritis (extracapillary glomerulonephritis with crescent formation). Nephrology (SaintPeters burg) 2015;19(6):73-82 (In Russ.)].
- Jennette JC. Rapidly progressive crescentic glomerulonephritis. Kidney Int 2003;63:1164–77.
- McAdoo SP, Pusey CD. Antiglomerular basement membrane disease. Semin Respir Crit Care Med 2018;39(4):494-503.
- Moiseev S, Cohen Tervaert JW, Arimura Y, et al. 2020 international consensus on ANCA testing beyond systemic vasculitis. Autoimmun Rev 2020;19(9):102618.
- Aydin Z, Turkmen K, Dede F, et al. Demographic, clinical and laboratory characteristics of rapidly progressive glomerulonephritis in Turkey: Turkish Society of Nephrology-Glomerular Diseases (TSN-GOLD) Working Group. Clin Exp Nephrol 2021;25(2):173-83.
- Козловская Н.Л., Прокопенко Е.И., Эмирова Х.М., Серикова С.Ю. Клинические рекомендации по диагностике и лечению атипичного гемолитикоуремического синдрома. Нефрология и диализ 2015;17(3):242-64 [Kozlov skaya NL, Prokopenko EI, Emirova KhM, Serikova SYu. Clinical guidelines for diagnosis and treatment of atypical hemolytic uremic syndrome. Nephrology and Dialysis 2015;17(3):242-64 (In Russ.)].
- Коротчаева Ю.В., Козловская Н.Л., Демьянова К.А. и др. Атипичный гемолитико-уремический синдром: клиническая картина, диагностика и лечение. Клин фармакол тер 2022;31(2):43-50 [Korotchaeva Yu, Kozlovskaya N, Demyanova K, et al. Atypical hemolytic-uremic syndrome: clinical presentation, diagnosis and treatment. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(2):43-50 (In Russ.)].
- Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 2017;91(3):539-51.
- Hanna RM, Henriksen K, Kalantar-Zadeh K, et al. Thrombotic microangiopathy syndromes - common ground and distinct frontiers. Adv Chronic Kidney Dis 2022;29(2):149-60.
- Scheen M, Adedjouma A, Esteve E, et al. Kidney disease in antiphospholipid antibody syndrome: Risk factors, pathophysiology and management. Autoimmun Rev 2022;21(5):103072.
- Domingues V, Chock EY, Dufrost V, et al. Increased risk of acute and chronic microvascular renal lesions associated with antiphospholipid antibodies in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Autoimmun Rev 2022;21(10):103158.
- Giannakakis K, Faraggiana T. Histopathology of lupus nephritis. Clin Rev Allergy Immunol 2011;40(3):170-80.
- Martis N, Jamme M, Bagnis-Isnard C, et al; French Reference Centre for Thrombotic Microangiopathies. Systemic autoimmune disorders associated with thrombotic microangiopathy: A cross-sectional analysis from the French National TMA registry: Systemic autoimmune disease-associated TMA. Eur J Intern Med 2021;93:78-86.
- Hosman IS, Kos I, Lamot L .Serum amyloid A in inflammatory rheumatic diseases: a compendious review of a renowned biomarker. Front Immunol 2021;11: 631299.
- Düzgün N. Amyloid A amyloidosis and systemic lupus erythematosus. Expert Rev Clin Immunol 2007;3(5):701-8.
- He X, Ning JP, Xu H, et al. Renal amyloidosis secondary to ANCA-associated vasculitis: a case report. Chin Med Sci J 2022 Jun 28. doi: 10.24920/003999.
- Рамеев В.В., Козловская Л.В., Рамеева А.С. и др. Анализ современной этиологии АА-амилоидоза и оценка влияния ее изменений на диагностику и подходы к лечению. Терапевтический архив 2021;93(6):672–8 [Rameev VV, Kozlovskaya LV, Rameeva AS, et al. The analysis of secondary AA-amyloidosis current etiology and its influence on the approaches for diagnosis and treatment. Terapevticheskii Arkhiv 2021;93(6):672–8 (In Russ.)].
- Obici L, Merlini G. Amyloidosis in autoinflammatory syndromes. Autoimmun Rev 2012;12(1):14-7.
- Mukhin NA, Kozlovskaya LV, Bogdanova MV, et al. Predictors of AA amyloidosis in familial Mediterranean fever. Rheumatol Int 2015;35(7):1257-61.
- O’Hara R, Murphy EP, Whitehead AS, et al. Acute-phase serum amyloid A production by rheumatoid arthritis synovial tissue. Arthritis Res 2000;2:142–8.
- Ravichandran S, Lachmann HJ, Wechalekar AD. Epidemiologic and survival trends in amyloidosis, 1987-2019. N Engl J Med 2020;382(16):1567-8.
- Mazzucchelli R, Almodovar-González R, Dieguez-Costa E, et al. Trends in amyloidosis in spondyloarthritis: results from the Spanish National Inpatient Registry over a 17-year period (1999-2015)-TREND-EspA study. RMD Open 2021;7(3):e001782.
- Akse-Onal V, Sağ E, Ozen S, et al. Decrease in the rate of secondary amyloidosis in Turkish children with FMF: are we doing better? Eur J Pediatr 2010;169(8): 971-4.
- Рамеев В.В., Моисеев С.В., Козловская Л.В. AA-амилоидоз при аутовоспалительных заболеваниях. Клин фармакол тер 2021;30(4):52-61 [Rameev V, Moiseev S, Kozlovskaya L. AA amyloidosis in autoinflammatory diseases. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(4):52-61 (In Russ.)].
- Bohle A, Mackensen-Haen S, von Gise H, et al. The consequences of tubulointerstitial changes for renal function in glomerulopathies. A morphometric and cytological analysis. Pathol Res Pract 1990;186:135–44.
- Krassanairawiwong K, Charoenpitakchai M, Supasyndh O, et al. Revised ISN/RPS 2018 classification of lupus renal pathology predict clinical remission. Int Urol Nephrol 2021;53(7):1391-8.
- Brix SR, Noriega M, Tennstedt P, et al. Development and validation of a renal risk score in ANCA-associated glomerulonephritis. Kidney Int 2018;94(6):1177-88.
- Barbour SJ, Coppo R, Zhang H, et al. Evaluating a new international risk-prediction tool in IgA nephropathy. JAMA Intern Med 2019;179(7):942-52.
- François H, Mariette X. Renal involvement in primary Sjogren syndrome. Nat Rev Nephrol 2016 Feb;12(2):82-93.
- Najafian B, Fogo AB, Lusco MA, Alpers CE. AJKD Atlas of Renal Pathology: IgG4-related tubulointerstitial nephritis. Am J Kidney Dis 2017;69(4):e19-20.
- Lusco MA, Fogo AB, Najafian B, Alpers CE. AJKD Atlas of Renal Pathology: Gouty nephropathy. Am J Kidney Dis 2017;69(1):e5-6.
- Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD Atlas of Renal Pathology: sarcoidosis. Am J Kidney Dis 2016;68(1):e5-6.
- Lusco MA, Fogo AB, Najafian B, Alpers CE. AJKD Atlas of Renal Pathology: Thrombotic microangiopathy. Am J Kidney Dis 2016;68(6):e33-4.
- Коханчук В.А., Скворцов А.В., Щеголева Е.М. и др. Роль биопсии почки в определении тактики ведения пациентов ревматологического отделения: ретроспективное исследование. Терапевтический архив 2022;94(6):763–8 [Kokhanchuk VA, Skvortsov AV, Shchegoleva EM, et al. Impact of kidney biopsy on the management of patients in the rheumatology department: retrospective study. Terapevticheskii Arkhiv 2022;94(6):763–8 (In Russ.)].
- Berden AE, Ferrario F, Hagen EC, et al. Histopathologic classification of ANCAassociated glomerulonephritis. J Am Soc Nephrol 2010 ;21(10):1628-36.
- Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Kidney Int 2021;100(4S):S1-276.
- Moroni G, Vercelloni PG, Quaglini S, et al. Changing patterns in clinical-histological presentation and renal outcome over the last five decades in a cohort of 499 patients with lupus nephritis. Ann Rheum Dis 2018;77:1318–25.
- Rhee RL, Hogan SL, Poulton CJ, et al. Trends in long-term outcomes among patients with antineutrophil cytoplasmic antibody-associated vasculitis with renal disease. Arthritis Rheumatol 2016;68(7):1711-20.
- van Daalen EE, Jennette JC, McAdoo SP, et al. Predicting outcome in patients with anti-GBM glomerulonephritis. Clin J Am Soc Nephrol 2018;13(1):63-72.
- Буланов Н.М., Козловская Н.Л., Тао Е.А. и др. Современные подходы к лечению АНЦА-ассоциированных васкулитов с поражением почек с позиций медицины, основанной на доказательствах. Клин фармакол тер 2020;29(4):72-84 [Bulanov N, Kozlovskaya N, Tao E, et al. Evidence-based treatment of ANCA-associated vasculitis with kidney involvement. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2020;29(4):72-84 (In Russ.)].
- Моисеев С.В., Новиков П.И., Буланов Н.М. Системная красная волчанка: эпидемиология, отдаленные исходы и бремя болезни. Клин фармакол тер 2021;30(4):13-22 [Moiseev S, Novikov P, Bulanov N. Systemic lupus erythematosus: epidemiology, outcomes and burden. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(4):13-22 (In Russ.)].
- Liossis SN, Staveri C. What's new in the treatment of systemic lupus erythematosus. Front Med (Lausanne) 2021;8:655100.
- Xiao H, Dairaghi DJ, Powers JP, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J Am Soc Nephrol 2014;25:225-31.
- Jayne DRW, Merkel PA, Schall TJ, Bekker P; ADVOCATE Study Group. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med 2021;384(7):599-609.
- Fernandez-Ruiz R, Belmont HM. The role of anticomplement therapy in lupus nephritis. Transl Res 2022;245:1-17.
- Саркисова И.А., Рамеев В.В., Козловская Л.В. Факторы риска развития и прогрессирования АА-амилоидоза у больных ревматоидным артритом. Нефрология и диализ 2007;9(3):346 [Sarkisova IA, Rameev VV, Kozlovskaya LV. Risk factors for development and progression of AA-amyloidosis in patients with rheumatoid arthritis. Nefrologiya i dializ 2007;9(3):346 (In Russ.)].
- Babaoglu H, Armagan B, Bodakci E, et al. Predictors of persistent inflammation in familial Mediterranean fever and association with damage. Rheumatology 2021;60:333-9.
- Okuda Y, Ohnishi M, Matoba K, et al. Comparison of the clinical utility of tocilizumab and anti-TNF therapy in AA amyloidosis complicating rheumatic diseases. Mod Rheumatol 2014;24(1):137-43.
- Pamuk ЕN, Kalyoncu U, Aksu K, et al. A multicenter report of biologic agents for the treatment of secondary amyloidosis in Turkish rheumatoid arthritis and ankylosing spondylitis patients. Rheumatol Int 2016;36(7):945-53.
- Lane T, Loeffler JM, Rowczenio DM, et al. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum 2013;65(4):1116-21.
- Tektonidou MG, Dasgupta A, Ward MM. Risk of end-stage renal disease in patients with lupus nephritis, 1971-2015: A systematic review and Bayesian metaanalysis. Arthritis Rheumatol 2016;68(6):1432-41.
- Lionaki S, Hogan SL, Jennette CE, et al. The clinical course of ANCA small-vessel vasculitis on chronic dialysis. Kidney Int 2009;76(6):644-51.
- Kalantar-Zadeh K, Jafar TH, Nitsch D, et al. Chronic kidney disease. Lancet 2021;398:786-802.
- Heerspink HJL, Stefánsson BV, Correa-Rotter R, et al.; DAPA-CKD Trial Committees and Investigators. Dapagliflozin in patients with chronic kidney disease. N Engl J Med 2020;383(15):1436-46.
- Кemann M, Kronbichler A. Call for action in ANCA-associated vasculitis and lupus nephritis: promises and challenges of SGLT-2 inhibitors. Ann Rheum Dis 2022;81(5):614-7.
- De Benedetti F, Gattorno M, Anton J, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med 2018;378:1908-19.