Острое или прогрессирующее ухудшение функции почек при иммуновоспалительных ревматических заболеваниях
Основные причины острого или прогрессирующего поражения почек при системных аутоиммунных заболеваниях включают в себя быстропрогрессирующий гломерулонефрит (АНЦА-ассоциированные васкулиты, анти-БМК болезнь, реже другие васкулиты и системная красная волчанка), тромботическую микроангиопатию (антифосфолипидный синдром, в том числе при системной красной волчанке, а также системная склеродермия, АНЦА-ассоцированные васкулиты и др.) и лекарственноиндуцированное острое повреждение почек (некроз канальцев, обусловленный нарушениями внутрипочечной гемодинамики или реже прямым токсическим действием лекарств, острый тубулоинтерстициальный нефрит). Важную роль в этиологии последнего играют нестероидные противовоспалительные препараты. Причинами внезапного ухудшения функции почек могут быть также васкулопатия почек при системной склеродермии (склеродермический почечный криз), острый тубулоинтерстициальный нефрит при синдроме Шегрена или IgG4-ассоциированной болезни, рабдомиолиз (миоглобинурия) при идиопатических воспалительных миопатиях, острая уратная нефропатия при подагре, тромбоз почечной артерии или вены при антифосфолипидном синдроме. Помимо клинических данных, важное значение в дифференциальной диагностике причин внезапного ухудшения азотвыделительной функции почек имеют результаты серологического исследования (АНЦА, антитела к базальной мембране клубочков, антифосфолипидные антитела, крио-глобулины, антинуклеарный фактор, антитела к ДНК и др.) и биопсии почек, которая часто необходима для выбора тактики лечения.
Поражение почек нередко развивается при любых системных аутоиммунных заболеваниях и многих аутовоспалительных заболеваниях и может привести к развитию хронической почечной недостаточности, требующей заместительной почечной терапии (диализ или трансплантация почки) [1]. Обычно азотвыделительная функция почек снижается постепенно под действием не только основного заболевания, но и других факторов, таких как артериальная гипертония или сахарный диабет. Однако возможно и быстрое ухудшение функции почек, когда лечение диализом приходится начинать уже через несколько дней или недель после появления или нарастания изменений в моче и повышения сывороточного уровня креатинина. Острое или быстро прогрессирующее ухудшение функции почек может быть обусловлено различными причинами, установление которых необходимо для выбора адекватного лечения, позволяющего восстановить функцию почек или по крайней мере отсрочить начало заместительной почечной терапии. Во многих случаях дифференциальная диагностика невозможна без биопсии почки, учитывая сходство лабораторных признаков нефропатий и возможность участия различных механизмов в развитии повреждения почек при одном и том же заболевании. Кроме того, при отсутствии результатов предыдущих исследований бывает сложно оценить длительность повышения сывороточного уровня креатинина, т.е. дифференцировать потенциально обратимое и необратимое поражение почек.
Основные понятия
Номенклатура нарушений функции и заболеваний почек была недавно пересмотрена на конференции KDIGO (Kidney Disease: Improving Global Outcomes) [2]. Классификация хронической болезни почек (ХБП), основанная на оценке расчетной скорости клубочковой фильтрации (СКФ) и альбуминурии, не претерпела изменений, однако эксперты не рекомендовали пользоваться термином "терминальная" применительно к почечной недостаточности или стадии ХБП, так как лечение диализом или трансплантация почки позволяют на многие годы продлить жизнь пациентам с ХБП 5 стадии. Более того, было рекомендовано считать "почечной недостаточностью" только снижение СКФ <15 мл/мин/1,73 м2 или необходимость в лечении диализом, в то время как на практике хроническую почечную недостаточность обычно устанавливают при снижении СКФ <60 мл/мин/1,73 м2. Быстрое ухудшение азотвыделительной функции наблюдается при остром повреждении (ОПП) или острой болезни почек (ОБП). Первое характеризуется внезапным развитием олигурии и увеличением сывороточного уровня креатинина более чем на 0,3 мг/дл в течение 2 дней или более чем на 50% в течение недели. ОПП рассматривают как вариант острой болезни почек (ОБП), которая включает в себя также состояния, сопровождающиеся менее выраженным или более медленным (в течение более 7 дней, но до 3 мес) развитием нарушения функции почек [3]. Номенклатура KDIGO не предполагает выделение быстропрогрессирующей почечной недостаточности (в отечественной литературе используется термин "быстропрогрессирующий нефритический синдром"), под которой понимают увеличение сывороточного уровня креатинина по крайней мере в два раза в течение ≤3 мес, характерное для быстропрогрессирующего гломерулонефрита (БПГН) [4]. По сути дела, "быстрое прогрессирование" укладывается в понятие ОБП, хотя с практической точки зрения следует отделять продолжающееся нарастание сывороточного уровня креатинина от персистирования признаков повреждения почек в течение более 7 дней, учитывая тяжесть течения и неблагоприятность прогноза БПГН. В отличие от ОПП, ОБП и ХБП, которые описывают нарушения функции и/или изменения структуры почек, БПГН является одним из вариантов гломерулонефрита. Как ОПП, так и ОБП далеко не всегда заканчиваются формированием ХБП, однако они тесно связаны между собой и являются компонентами единого континуума (рис. 1).
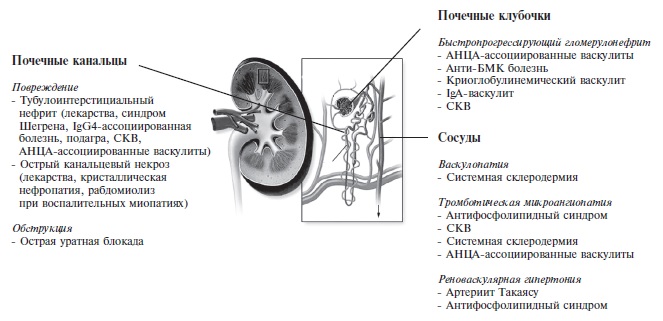
В данной статье рассматриваются причины любого внезапного или прогрессирующего ухудшения функции почек, которое обусловлено активным почечным процессом, т.е. не отражает естественную эволюцию ХБП, связанную с постепенным нарастанием фиброза почек. Помимо темпов ухудшения функции почек, доводами в пользу активности заболевания обычно служат изменения в моче (микрогематурия, протеинурия), а также отсутствие признаков сморщивания почек при ультразвуковом или рентгенологическом исследовании. При иммуновоспалительных ревматических заболеваниях причиной нарушения функции почек может быть повреждение любых структур нефрона, в том числе клубочков (БПГН при системных васкулитах и других заболеваниях), канальцев (лекарства, болезнь Шегрена, IgG4-ассоциированная болезнь и др.) и сосудов (васкулопатия при системной склеродермии или тромботическая микроангиопатия [ТМА] при антифосфолипидном синдроме [АФС]) (рис. 2).
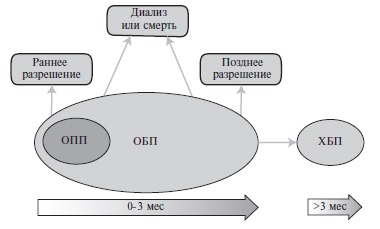
В данной статье рассматриваются причины любого внезапного или прогрессирующего ухудшения функции почек, которое обусловлено активным почечным процессом, т.е. не отражает естественную эволюцию ХБП, связанную с постепенным нарастанием фиброза почек. Помимо темпов ухудшения функции почек, доводами в пользу активности заболевания обычно служат изменения в моче (микрогематурия, протеинурия), а также отсутствие признаков сморщивания почек при ультразвуковом или рентгенологическом исследовании. При иммуновоспалительных ревматических заболеваниях причиной нарушения функции почек может быть повреждение любых структур нефрона, в том числе клубочков (БПГН при системных васкулитах и других заболеваниях), канальцев (лекарства, болезнь Шегрена, IgG4-ассоциированная болезнь и др.) и сосудов (васкулопатия при системной склеродермии или тромботическая микроангиопатия [ТМА] при антифосфолипидном синдроме [АФС]) (рис. 2).
Быстропрогрессирующий гломерулонефрит
БПГН – наиболее тяжелый вариант гломерулонефрита, требующий неотложного назначения иммуносупрессивной терапии, которая позволяет затормозить прогрессирование почечной недостаточности или восстановить функцию почек [4]. Характеризуется наличием острого нефритического синдрома и быстрым нарастанием сывороточного уровня креатинина по крайней мере в два раза в течение ≤3 мес (быстропрогрессирующий нефритический синдром). У части больных темпы снижения функции почек соответствуют критериям ОПП, а лечение диализом приходится начинать уже через несколько дней после появления первых симптомов заболевания.
Основными причинами БПГН являются АНЦАассоциированные васкулиты (ААВ) и болезнь, связанная с антителами к базальной мембране клубочков (анти-БМК), более редкими – криоглобулинемический васкулит, IgA-васкулит (старое название – геморрагический васкулит или пурпура Шейнлейн-Геноха) и системная красная волчанка (СКВ). Для подтверждения диагноза и установления причины БПГН всем больным рекомендуются биопсия почек и определение специфических антител (АНЦА, анти-БМК, криоглобулины, антинуклеарные антитела). При гистологическом исследовании нефробиоптата выявляют экстракапиллярный гломерулонефрит с клеточными или фиброзно-клеточными полулуниями по крайней мере в 50% клубочков. В зависимости от типа свечения при иммунофлюоресцентном исследовании и серологического профиля выделяют три типа БПГН: I – связанный с антителами к базальной мембране клубочков (анти-БМК болезнь), II – иммунокомплексный (СКВ, криоглобулинический васкулит и др.) и III – малоиммунный (ААВ).
АНЦА-ассоциированные васкулиты. В американском исследовании у 60% из 632 пациентов с БПГН, подтвержденным при биопсии почки, был выявлен малоиммунный экстракапиллярный гломерулонефрит, основной причиной которого являются ААВ [5]. Среди больных старше 60 лет доля его в структуре БПГН достигла 79%, что отражает особенности эпидемиологии ААВ, который чаще развивается у людей старшего и пожилого возраста. В турецком исследовании ААВ также был диагностирован примерно у половины из 200 больных БПГН [6]. В нашем исследовании частота быстропрогрессирующего нефритического синдрома среди 374 больных ААВ составила 19% [7]. Она была выше у больных микроскопическим полиангиитом (58%) и значительно ниже у пациентов с гранулематозом с полиангиитом и эозинофильным гранулематозом с полиангиитом (12% и 4%, соответственно). Пред полагать ААВ у пациентов с БПГН следует при наличии АНЦА (чаще к миелопероксидазе; иногда антитела не определяются, если иммуносупрессивная терапия была начата до иммуноферментного анализа) и системных проявлений, хотя последние могут и отсутствовать. Среди системных проявлений наиболее опасным является диффузное альвеолярное кровотечение, которое чаще развивается у пациентов с микроскопическим полиангиитом, проявляется кровохарканьем, анемией и диффузными интерстициальными изменениями легких и может привести к развитию острого респираторного дистресс-синдрома [8]. В отличие от микроскопического полиангиита, у пациентов с гранулематозом с полиангиитом поражение почек обычно сочетается с признаками гранулематозного воспаления легких и/или верхних дыхательных путей, а у больных эозинофильным гранулематозом с полиангиитом – с эозинофильной бронхиальной астмой/риносинуситом.
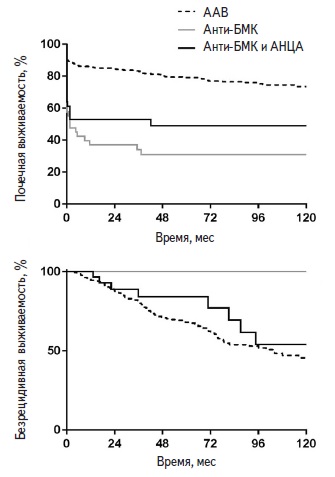
Анти-БМК болезнь (старое название – синдром Гуд пас чера). В американской когорте доля экстракапиллярного гломерулонефрита I типа, характеризующегося циркуляцией анти-БМК и линейным свечением антител в почечном биоптате, в структуре БПГН составила 15% и не отличалась у пациентов молодого и пожилого возраста [5]. Анти-БМК болезнь – это системный васкулит, сопровождающийся образованием аутоантител к антигенам коллагена IV типа, который является компонентом базальных мембран почечных клубочков и альвеол [9]. БПГН развивается практически у всех больных с анти-БМК болезнью и примерно в 40% случаев сочетается с диффузным альвеолярным кровотечением. Как указано выше, помимо анти-БМК болезни причинами легочно-почечного синдрома могут быть микроскопический полиангиит и гранулематоз с полиангиитом. БПГН при анти-БМК болезни характеризуется очень быстрым ухудшением функции почек (по типу ОПП), а в когортных исследованиях около 60% пациентов начинали лечение гемодиализом до установления диагноза [9]. В отличие от ААВ, для анти-БМК не характерны рецидивы, однако почечная выживаемость несмотря на агрессивную иммуносупрессивную терапию ниже (4050% в течение 1 года) [10,11]. В клинических исследованиях примерно у трети больных с анти-БМК болезнью определялись не только анти-БМК, но и АНЦА [9] Клинический фенотип у пациентов с двойной позитивностью сходен с таковым анти-БМК болезни без АНЦА, включая частое развитие тяжелого поражения почек и легочного кровотечения в начале заболевания, хотя у них определяются и некоторые черты, свойственные ААВ, такие как пожилой возраст, наличие внепочечных проявлений помимо поражения легких, более высокая вероятность восстановления функции почек и риск рецидивов [12]. В международном исследовании рецидивы развивались примерно у половины пациентов с ААВ и анти-БМК болезнью с двойной позитивностьюи отсутствовали у всех больных, у которых определя-лись только анти-БМК (рис. 3) [13]. При этом доляпациентов с диализ-зависимой почечной недостаточ-ностью, у которых через 1 год восстановилась функцияпочек, среди больных с анти-БМК болезнью, у которыхциркулировали только анти-БМК или антитела двухтипов, составила, соответственно, 17% и 29%, а средибольных ААВ – 49%.
Другие заболевания. Другие системные васкулиты, в том числе криоглобулинемический и IgA-васкулит, и СКВ являются редкими причинами быстропрогрессирущего нефритического синдрома, хотя доля иммунокомплексного гломерулонефрита II типа, развивающегося при этих заболеваниях, в структуре БПГН в американском исследовании составила 24% [5]. Она была выше у детей и подростков (45%) и людей в возрасте от 21 до 60 лет (35%) и значительно ниже у пациентов старше 60 лет (6%). Частой причиной этого варианта БПГН служит стрептококковая инфекция. Предполагать указанные заболевания у пациентов с быстро прогрессирующим ухудшением функции почек следует при наличии характерных клинических и лабораторных проявлений. Например, волчаночный нефрит чаще встречается у молодых женщин, характеризуется развитием не только нефритического, но и нефротического синдрома, сопровождается типичными системными проявлениями (артрит, кожная сыпь, цитопения, гипокомплементемия и т.п.) и наличием соответствующих аутоантител (антинуклеарный фактор, антитела к дсДНК, Smith антигену и др.). Для криоглобулинемического васкулита характерно наличие HCV- или реже HBV-инфекции (хотя иногда васкулит развивается у неинфицированных пациентов) и криоглобулинов в сыворотке крови, а для IgA-васкулита – рецидивирующей кожной пурпуры и артрита, обычно появляющихся в молодом возрасте.
Лечение. Тактика лечения БПГН в целом сходная при различных системных аутоиммунных заболеваниях ипредполагает сочетанное применение глюкокортикостероидов, в том числе в сверхвысоких дозах, и иммуносупрессивных препаратов, прежде всего циклофосфамида.Однако рекомендуемый режим дозирования (а в случаеанти-БМК болезни и путь введения) этих препаратовотличается в зависимости от нозологической формы.При ААВ, криоглобулинемическом васкулите и IgAваскулите назначают циклофосфамид внутривенно вдозе 15 мг/кг (но не более 1200 мг) каждые 2 недели, азатем 3 недели (не менее 6 введений), при этом дозуследует корректировать по возрасту и величине СКФ[14]. Быстропрогрессирующее течение волчаночногонефрита может быть основанием для выбора болеевысоких, чем традиционно (500 мг внутривенно №6 синтервалом в 2 недели), доз циклофосфамида – 0,5–0,75 г/м2 ежемесячно [15]. Единственным заболеванием,при котором предпочтительным остается пероральныйприем циклофосфамида, является анти-БМК болезнь,при которой препарат применяют в дозе 2 мг/кг/сут (ноне более 200 мг) на протяжении трех месяцев [14].Кроме того, переход на пероральный прием циклофосфамида возможен для преодоления резистентного течения ААВ [16]. Следует отметить, что доказательная базапо применению ритуксимаба у пациентов с аутоиммунными заболеваниями с тяжелым поражением почек внастоящее время недостаточна для окончательного подтверждения его эквивалентности циклофосфамиду,поэтому во многих рекомендациях ритуксимаб рассматривается как компонент терапии второй линии.
В последние годы претерпели изменения подходы к терапии глюкокортикостероидами с тенденцией к быстрому снижению дозы препарата и уменьшению продолжительности лечения. Лечение пациентов с БПГН традиционно начинают с одного-трех сеансов пульс-терапии глюкокортикостероидами, хотя преимущество ее перед пероральным приемом препарата убедительно не доказано. Затем пациенты получают глюкортикостероиды внутрь в начальной дозе, не превышающей 1 мг/кг (но не более 75 мг/сут). В настоящее время наблюдается тенденция к уменьшению кумулятивной дозы глюкокортикостероидов за счет быстрых темпов снижения дозы (как при ААВ) и/или уменьшения начальной дозы препарата до 0,5 мг/кг (как при СКВ) [15,17]. При волчаночном нефрите присоединение к стандартной терапии белимумаба повышает вероятность достижения ремиссии [18].
Открытым остается вопрос о роли плазмообмена в лечении БПГН. В то время как плазмообмен является обязательным компонентом терапии анти-БМК болезни, значимо влияющим на прогноз для жизни, его эффективность при других формах БПГН ясна не в полной мере. В частности, в наиболее крупном исследовании эффективности плазмообмена при ААВ не было подтверждено влияние этого метода лечения на вероятность наступления комбинированной конечной точки (смерть или хроническая почечная недостаточность в течение 12 мес) [17]. В то же время последующие метаанализы рандомизированных клинических исследований показали потенциальное положительное влияние плазмообмена на почечную выживаемость пациентов с ААВ в краткосрочной перспективе [19,20].
После достижения ремиссии заболевания пациенты на протяжении не менее 1,5-2 лет получают поддерживающую терапию глюкокортикостероидами в низкой дозе и менее токсичными, чем циклофосфамид, иммуносупрессивными препаратами, такими как ритуксимаб, азатиоприн или микофенолата мофетил. Единственным исключением является БПГН в рамках анти-БМК болезни, при которой поддерживающую терапию не проводят ввиду крайне низкого риска обострений.
Тромботическая микроангиопатия
ТМА – это синдром, развивающийся в результате повреждения эндотелия и образования тромбов в сосудах микроциркуляторного русла, что приводит к их окклюзии и ишемии органов и тканей, прежде всего почек [21]. Характерные проявления ТМА включают в себя ОПП, тромбоцитопению и микроангиопатическую гемолитическую анемию. Помимо анемии на внутрисосудистый гемолиз указывают отрицательная проба Кумбса и увеличение активности ЛДГ, а также повышение количества фрагментированных эритроцитов (шистоцитов) более >0,1% и снижение содержания гаптоглобина [22]. При биопсии почки у пациентов с острой ТМА обнаруживают микротромбы в почечных артериолах и/или клубочках при отсутствии признаков воспаления и иммунных депозитов при иммунофлюоресцентом исследовании [23]. У пациентов с СКВ эти изменения могут сочетаться с признаками волчаноч ного нефрита. При СКВ важное диагностическое значение имеет биопсия почки, которая является единственным методом, позволяющим надежно дифференцировать ухудшение функции почек, связанное с ТМА или БПГН, и выбрать адекватное лечение (антикоагулянты или иммуносупрессивные препараты, соответственно).
К первичной ТМА относят тромботическую тромбоцитопеническую пурпуру, типичный гемолитико-уремический синдром (STEC-ГУС) и атипичный ГУС, в то время как вторичная ТМА развивается под действием различных факторов, в том числе лекарств, инфекций, опухолей, беременности, трансплантации органов [24]. В ревматологии основной причиной ТМА является антифосфолипидный синдром (АФС), в том числе при СКВ, более редкими – системная склеродермия, ААВ и другие аутоиммунные заболевания, а также нежелательные эффекты медикаментозной терапии, в частности циклоспорином. Для исключения АФС у больных с ОПП необходимо определять антифосфолипидные антитела, в том числе волчаночный антикоагулянт (исследование не имеет смысла на фоне терапии антикоагулянтами), IgG и IgM антитела к кардиолипину и β2-гликопротеину, особенно при наличии указаний на артериальные и/или венозные тромбозы (исключая тромбоз поверхностных вен) и акушерские осложнения (необъяснимая смерть плода на сроке более 10 недель, преждевременные роды на сроке до 34 недель вследствие эклампсии или плацентарной недостаточности и/или по крайней мере три спонтанных аборта на сроке до 10 недель) в анамнезе [25]. Нефропатию не относят к критериям диагностики АФС, диагноз которого у пациентов с антифосфолипидными антителами может быть установлен только при наличии тромбозов и/или акушерских осложнений. Однако даже при отсутствии указанных клинических критериев АФС может быть диагностирован, если при нефробиопсии определяются микротромбы в артериолах или клубочках почек, а в крови циркулируют антифосфолипидные антитела.
Помимо образования микротромбов в артериолах, характерного для ТМА, при АФС возможно и поражение крупных сосудов почек с развитием одно- или двустороннего стеноза или тромбоза почечных артерий, сопровождающегося ишемической нефропатией и вторичной артериальной гипертонией, тромбоза почечной вены и инфаркта почки [23]. ОПП может наблюдаться в рамках катастрофического АФС, который представляет собой генерализованную ТМА и характеризуется поражением не только почек, но и других органов (центральной нервной системы, сердца, легких и др.) с развитием полиорганной недостаточности. В связи с этим у пациентов с АФС возможно как фульминантное развитие почечной недостаточности, так и постепенное ухудшение функции почек.
Данные об эпидемиологии нефропатии при АФС ограничены. В наиболее крупном исследовании среди 1000 пациентов с АФС (у 36% из них имелась СКВ) признаки поражения почек (тромбоз клубочков, инфаркт почки, тромбоз почечной артерии или вены) были выявлены в 2,7% случаев [26]. В той же когорте развитие катастрофического АФС, проявления которого, как правило, включают в себя ОПП, в течение 10 лет было зарегистрировано у 0,9% пациентов [27]. Следует учитывать, что даже катастрофический АФС не всегда сопровождается типичными лабораторными признаками ТМА. I. Rodriguez-Pintó и соавт. проанализировали 522 случая катастрофического АФС у 500 больных (у 40% имелись сочетанные аутоиммунные заболевания, прежде всего СКВ) [28]. Нарушение функции почек отмечено в 77% случаев. Частота ТМА, проявлявшейся сочетанием тромбоцитопении, гемолитической анемии и увеличением количества шистоцитов, составила всего 14%, хотя "изолированная" тромбоцитопения была выявлена у 67% больных, а повышенное число шистоцитов – у 22%.
С другой стороны, у пациентов с СКВ образование микротромбов в почках может наблюдаться при отсутствии циркулирующих антифосфолипидных антител. V. Domingues и соавт. провели мета-анализ 35 исследований, в которых у 3035 пациентов с СКВ оценивали частоту ТМА на основании гистологического исследования биоптатов почки [29]. Признаки острой или хронической ТМА были выявлены у 454 больных, а частота ее у пациентов, у которых определялись и не определялись антифосфолипидные антитела, составила 31,3% и 10,4%, соответственно. Результаты этого исследования служат дополнительным доводом в пользу биопсии почки с целью дифференциальной диагностики различных причин ОПП у пациентов с СКВ.
Развитие АФС наблюдается не только при СКВ, но и при других ревматических иммуновоспалительных заболеваниях. В упомянутом выше исследовании у 2,2% из 1000 пациентов с АФС был диагностирован синдром Шегрена, у 1,8% – ревматоидный артрит, у 0,7% – системная склеродермия, у 0,7% – системный васкулит, у 0,5% – дерматомиозит [26]. По данным систематизированного обзора, частота выявления антифосфолипидных антител, подтвержденного при повторном анализе через 12 недель, при различных ревматических заболеваниях (системной склеродермии, ревматоидном артрите, синдроме Шегрена и др.), варьировалась от 0 до 30% (медиана 11%) [30]. Чаще всего эти антитела определялись у пациентов с системной склеродермией.
В лечении поражения почек у пациентов с АФС ключевую роль играет не иммуносупрессивная, а антикоагулянтная терапия гепарином с последующей заменой на антагонисты витамина К для вторичной профилактики тромбозов [31]. Развитие ТМА может быть показанием для проведения плазмообмена с эксфузией 40-60 мл/кг и восполнением объема альбумином и/или свежезамороженной плазмой. В качестве терапии спасения описано применение экулизумаба, однако его эффект наиболее выражен при состояниях, сопровождающихся активацией комплемента по альтернативному пути [32].
Склеродермический почечный криз
Склеродермический почечный криз (СПК), впервые описанный более 150 лет назад, характеризуется внезапным повышением АД и развитием ОПП, которое примерно в половине случаев сочетается с признаками ТМА [33,34]. В прошлом частота СПК при диффузной форме системной склеродермии достигала 25%, однако в последующем она снизилась [35]. По данным метаанализа клинических исследований, опубликованного в 2016 г., распространенность СПК во всей популяции пациентов с системной склеродермией составила 4%, но была несколько выше при диффузной форме болезни – 7-9% [36]. Среди 9690 пациентов с системной склеродермией, включенных в базу данных EUSTAR, развитие СПК было отмечено в 169 случаях (2%), а заболеваемость составила всего 3,75 на 1000 пациентов в год [37]. Возможными причинами снижения частоты СПК считают широкое применение вазодилататоров для лечения синдрома Рейно и легочной артериальной гипертонии и более осторожное назначение глюкокортикостероидов, которые повышают риск развития СПК у пациентов с системной склеродермией [35]. M. Pes taña-Fernández и соавт. в ретроспективном исследовании у 544 пациентов с системной склеродермией, осложнившейся дигитальными язвами, выявили увели че ние срока до развития СПК на фоне лечения антагонистами эндотелина или ингибиторами фосфодиэстеразы 5-го типа в среднем с 2,9 до 7,6 лет, хотя частота СПК была сходной в группах больных, получавших и не получавших препараты этих групп (3,4 и 2,7 на 1000 пациентов в год, соответственно) [38].
Причиной развития СПК считают повреждение сосудистых эндотелиальных клеток, которое сопровождается выделением факторов роста и цитокинов, вызывающих пролиферацию гладкомышечных клеток, гиперплазию и фиброз интимы внутрипочечных сосудов [33,35]. Пролиферативная васкулопатия приводит к ишемии клубочков и стойкой активации ренин-ангиотензин-альдостероновой системы. Роль последней в патогенезе СПК подтверждается эффективностью ингибиторов АПФ при этом состоянии, хотя не исключается значение и других факторов, таких как спазм почечных артериол ("почечный" синдром Рейно). Таким образом, СПК не относится к аутоиммунным проявлениям системной склеродермии, хотя аутоантитела, прежде всего к РНК-полимеразе III, могут быть триггером активации эндотелия [35]. Высокая частота ТМА у пациентов с почечной васкулопатией может указывать на роль активации системы комплемента в патогенезе СПК, однако этот вопрос нуждается в дополнительном изучении [33].
Недавно эксперты рабочей группы Scleroderma Clinical Trials Consortium выделили основные проявления СПК, которые предполагается использовать для разработки классификационных критериев [39]:- Внезапное повышение АД (≥140/90 мм рт. ст. или по крайней мере на 30/20 мм рт. ст. выше обычного).
- ОПП.
- Микроангиопатическая анемия и тромбоцитопения (появление или нарастание анемии, шистоциты в мазке крови, тромбоцитопения ≤100000 в мм3, лабораторные признаки гемолиза, включая повышение активности ЛДГ, ретикулоцитоз и/или снижение уровня гемоглобина, отрицательная проба Кумбса).
- Нарушение функции органов-мишеней (гипертоническая ретинопатия и/или энцефалопатия, острая сердечная недостаточность, острый перикардит).
- Гистологические изменения в биоптате почки (преимущественное поражение артериол, признаки ТМА, неспецифические проявления ишемии, сужение и облитерация сосудов и др.).
Одно из основных проявлений СПК – умеренная или тяжелая артериальная гипертония, которая может осложниться развитием ретинопатии, нарушений мозгового кровообращения и/или острой сердечной недостаточности, однако у 10% пациентов с СПК увеличение АД отсутствует (нормотензивный вариант криза). По мнению Н. Yamashita и соавт., с патофизиологической точки зрения следует выделять два типа СПК, которые характеризуются сходными проявлениями, но развиваются в различной последовательности: васкулопатию, вызывающую утолщение интимы и артериальную гипертонию, и ТМА, сопровождающуюся повреждением стенок сосудов с развитием микрососудистого тромбоза [40]. При первом варианте СПК первоначально повышаются АД и сывороточный уровень креатинина, а тромбоцитопения присоединяется позднее и менее выражена, в то время как при втором варианте отмечается ранняя и тяжелая тромбоцитопения, а затем развиваются артериальная гипертония и ОПП.
СПК чаще встречается у пациентов с диффузной формой системной склеродермии (обычно в первые 1-4 года после начала заболевания), у которых нередко наблюдаются шум трения сухожилий, полиартрит, плотный отек кистей и/или синдром карпального канала, позволяющие предсказать прогрессирование заболевания [35]. Среди серологических маркеров системной склеродермии наиболее важное значение имеет наличие антител к РНК полимеразе III, которое в исследовании EUSTAR у 2800 пациентов ассоциировалось с 6-кратным увеличением риска развития СПК [41]. Лечение глюкокортикостероидами, особенно в более высоких дозах (более 15 мг/сут), также увеличивает риск СПК, в связи с чем целесообразно избегать их назначения пациентам с диффузной формой склеродермии. Хотя ингибиторы АПФ представляют собой основу лечения СПК, в крупном исследовании у 6521 пациента с системной склеродермией их применение с антигипертензивной целью, в отличие от антагонистов кальция, блокаторов ангиотензиновых рецепторов и антагонистов эндотелиновых рецепторов, ассоциировалось с увеличением отношения рисков этого осложнения в 2,55 раза (95% доверительный интервал 1,65-3,95) (рис. 4) [37]. В том же исследовании не было выявлено связи между лечением глюкокортикостероидами и СПК, однако во всей выборке доля пациентов, получавших эти препараты в дозе более 15 мг/сут в пересчете на преднизолон, составляла всего 3%.
![Кумулятивная частота СПК у пациентов, получавших и не получавших ингибиторы АПФ [37]](/wp-content/uploads/2023/04/ostroe-ili-progressiruyushchee-uhudshenie-funkcii-pochek-pri-immunovospalitelnyh-revmaticheskih-zabolevaniyah_fig34jpg.jpg)
Внедрение ингибиторов АПФ для лечения СПК привело к значительному улучшению прогноза больных с этим осложнением системной склеродермии. Тем не менее, по данным недавного опубликованного систематизированного обзора 20 исследований более чем у 14000 пациентов с системной склеродермией, смертность больных СПК составляет около 20% через 6 мес, 30-36% через 1 год и 19-40% через 3 года [42]. Хотя у части пациентов происходит восстановление функции почек (в течение до 2-3 лет), от 19 до 40% пациентов, перенесших СПК, нуждаются в постоянной заместительной почечной терапии. При этом трансплантация почки сопровождается более высокой выживаемостью больных по сравнению с таковой на фоне лечения диализом.
Тактика лечения СПК основывается на подавлении активности ренин-ангиотензин-альдостероновой системы ингибиторами АПФ (нередко каптоприлом), назначение которых многократно снижает риск летального исхода [43]. Антагонисты ангиотензиновых рецепторов по эффективности уступают ингибиторам АПФ. Возможно, это связано с тем, что они не ингибируют разрушение брадикинина, который оказывает сосудорасширяющее действие [35]. При недостаточной антигипертензивной эффективности ингибиторов АПФ к лечению добавляют другие препараты, в частности антагонисты кальция и доксазозин. Кроме того, эффективным может быть назначение препаратов простагландинов, обладающих вазодилатирующим эффектом. У пациентов с клиническими проявлениями ТМА лечение может быть усилено процедурами плазмообмена. Перспективным представляется также использование антагонистов эндотелина-1 для более быстрой нормализации почечного кровотока и АД [44], однако эффективность этих препаратов у пациентов с СПК не доказана.
Лекарственно-индуцированное ОПП
Лекарственные средства являются нередкой причиной развития ОПП у госпитализированных пациентов, особенно в отделениях реанимации и интенсивной терапии (ОРИТ). Например, в международном многоцентровом исследовании признаки ОПП были выявлены более чем у половины из 1082 пациентов, находившихся в ОРИТ, а доля нефротоксичных препаратов в его этиологии составила 14% [45]. Основной причиной ОПП при приеме лекарственных веществ является их прямое повреждающее действие на канальцы, хотя нарушение функции почек может быть обусловлено и другими механизмами [46-48]. Обсуждение всех групп лекарственных средств, которые могут вызвать ОПП, выходит за рамки настоящей статьи, поэтому ниже рассматриваются варианты повреждения почек при применении препаратов, предназначенных для лечения иммуновоспалительных ревматических заболеваний.
"Псевдо" ОПП. Повышение сывороточного уровня креатинина может имитировать ОПП при лечении некоторыми лекарственными средства, снижающими экскрецию креатинина в почечных канальцах (циметидин, триметоприм и др.) [47]. Развитие "псевдо" ОПП возможно также при лечении дексаметазоном, так как креатинин входит в состав его лекарственных форм в качестве наполнителя. Кроме того, все глюкокортикостероиды дают катаболический эффект и вызывают выделение из мышечной ткани креатина, превращающегося в креатинин.
Нарушение внутрипочечной гемодинамики. Препараты, оказывающие действие на внутрипочечную гемодинамику, могут вызвать ухудшение функции почек, обычно преходящее, за счет изменения кровотока по афферентной и/или эфферентной артериолам клубочка [47]. Этот эффект характерен для ингибиторов АПФ и блокаторов ангиотензиновых рецепторов, которые вызывают преимущественное расширение эфферентной артериолы и снижают давление в клубочках. Нестероидные противовоспалительные препараты (НПВП) и ингибиторы кальциневрина могут быть причиной снижения СКФ за счет сужения эфферентной артериолы, в частности в результате подавления секреции простагландинов, обладающих вазодилатирующими свойствами. Значи тельное ухудшение перфузии почек может осложниться ишемическим некрозом канальцев. Факторами риска более выраженных нарушений внутрипочечной гемодинамики являются уменьшение объема циркулирующей крови, артериальная гипотония, пожилой возраст, сердечная недостаточность, нефротический синдром.
Некроз канальцев. Помимо ишемии почек причиной острого повреждения канальцев может быть прямое токсическое действие некоторых лекарственных веществ (аминогликозиды, ванкомицин, рентгеноконтрастные средства, содержащие йод, и др.). Немно гочисленные случаи острого некроза канальцев описаны при применении бисфосфонатов, прежде всего при внутривенном введении золендроновой и памидроновой кислот (последняя чаще вызывала развитие коллабирующего фокального сегментарного гломерулосклероза) в более высоких дозах в онкологии [49]. При лечении остеопороза риск нефротоксичности бисфосфонатов низкий [50]. Нефротоксичность, в том числе острая, является одним из основных факторов, ограничивающих применение циклоспорина А и других ингибиторов кальциневрина. Повреждение канальцев при применении этих препаратов (как и НПВП) в первую очередь связано с нарушением внутрипочечной гемодинамики вследствие увеличения активности симпатической и ренин-ангиотензиновой системы, сек реции эндотелина-1 и снижения продукции простагландинов и оксида азота [51].
Острый тубулоинтерстициальный нефрит. Лекар ствен ные средства (ингибиторы протонной помпы, НПВП, антибиотики) – основная причина (более 70% случаев) развития острого тубулоинтерстициального нефрита, который характеризуется инфильтрацией интерстиция почек воспалительными клетками и нередко сопровождается ОПП, а также клиническими проявлениями аллергии, такими как лихорадка, кожная сыпь и эозинофилия. При биопсии почки признаки острого тубулоинтерстициального нефрита обнаруживают у 5-27% пациентов с ОПП [52]. При лечении иммуновоспалительных ревматических заболеваний острый тубулоинтерстициальный нефрит чаще всего встречается при применении НПВП. Необходимо учитывать, что изменения в моче могут появиться через несколько недель или месяцев после начала лечения НПВП, а неспецифичность этих изменений и отсутствие других проявлений аллергии в значительной степени затрудняют дифференциальную диагностику с другими причинами ОПП. В связи с этим для подтверждения диагноза обычно требуется биопсия почки. Развитие острого тубулоинтерстициального нефрита возможно также при лечении аллопуринолом. В отличие от других форм лекарственно-индуцированного ОПП, для лечения острого тубулоинтерстициального нефрита применяют глюкокортикостероиды.
Кристаллические нефропатии. При применении некоторых лекарственных средств, в частности метотрексата в больших дозах, возможно образование кристаллов самих препаратов и их метаболитов в моче и развитие ОПП вследствие прямого токсического действия кристаллов, воспаления и обструкции канальцев [47]. У пациентов с кристаллической нефропатией в моче могут быть обнаружены цилиндры, содержащие кристаллы, что имеет важное диагностическое значение.
Другие причины
Причинами ОПП в ревматологии могут быть и некоторые другие заболевания, которые обсуждаются ниже.
Рабдомиолиз (миоглобинурия). Развитие ОПП может быть следствием рабдомиолиза, наблюдающегося у пациентов с идиопатическими воспалительными миопатиями [53]. Разрушение мышечной ткани при миозите сопровождается высвобождением миоглобина, который образует преципитаты, вызывающие обструкцию и повреждение канальцев почек. Во французском исследовании частота ОПП у 150 пациентов с дерматомиозитом, полимиозитом или антисинтетазным синдромом составила 10,7% [54]. Основными причинами его были лекарства и миоглобинурия. Характерный признак миоглобинурии – потемнение мочи. В отличие от макрогематурии, в осадке мочи отсутствуют эритроциты, но определяются пигментированные цилиндры.
Реноваскулярная гипертония. Сужение почечных артерий наблюдается при артериите Такаясу и АФС. Реноваскулярная гипертония вызывает хроническую ишемию почек, однако резкое снижение АД при двустороннем стенозе почечных артерий может осложниться ОПП. Этот эффект характерен для ингибиторов АПФ, которые снижают давление в клубочках за счет преимущественного расширения эфферентных артериол почек. При АФС причиной ОПП может быть также тромбоз почечной артерии или вены.
Острый тубулоинтерстициальный нефрит. Повреж дение канальцев нередко наблюдается у пациентов с иммуновоспалительными ревматическими заболеваниями, в частности СКВ и АНЦА-ассоциированным васкулитом, и может вносить вклад в развитие ХБП. При некоторых заболеваниях, прежде всего синдроме Шегрена, IgG4-ассоциированной болезни и саркоидозе, тубулоинтерстициальный нефрит является ведущим вариантом поражения почек [55-57]. Чаще он характеризуется хроническим течением, но иногда приводит и к развитию ОПП. A. Muriithi и соавт. проанализировали 133 случая острого тубулоинтерстициального нефрита, сопровождавшегося ОПП и подтвержденного при биопсии почки [58]. У 70% больных причиной поражения почек были лекарственные препараты (антибиотики, ингибиторы протонной помпы и НПВП), у 20% – аутоиммунные заболевания, прежде всего саркоидоз (9%), синдром Шегрена (4%) и IgG4-ассоциированная бо лезнь (2%). Сходные данные приводят и японские авторы, которые изучили 153 случая тубулоинтерстициального нефрита, подтвержденного при биопсии почки [59]. Признаки ОПП на момент биопсии имелись у 107 больных. IgG4-ассоциированная болезнь была причиной поражения почек у 9,4% больных, синдром Шегрена – у 5,0% и саркоидоз – у 4,3%. Примерно у 70% пациентов с аутоиммунными заболеваниями ОПП отсутствовало или отмечалось ОПП 1 стадии, в то время как у большинства больных лекарственным тубулоинтерстициальным нефритом наблюдалось ОПП 2-3 стадии.
Острая уратная нефропатия (мочекислая блокада). У пациентов с подагрой причиной ОПП может быть быстрое нарастание урикемии и образование кристаллов мочевой кислоты, вызывающих обструкцию и повреждение почечных канальцев [60]. Прово циру ющи ми факторами могут быть дегидратация (например, после обильного потоотделения в бане), употребление пищевых продуктов, содержащих большое количество пуриновых оснований, и алкоголя, интенсивные физические нагрузки. Характерные симптомы – олигурия, повышение АД и выделение бурой мочи.
Заключение
Быстрое ухудшение функции почек (ОБП) при иммуновоспалительных ревматических заболеваниях может быть вызвано различными причинами, включая БПГН, ТМА, некроз почечных канальцев, острый тубулоинтерстициальный нефрит, кристаллические нефропатии и др. ОПП чаще всего развивается под действием лекарственных средств (например, НПВП), которые вызывают некроз почечных канальцев или острый тубулоинтерстициальный нефрит. Однако ОПП может бытьи проявлением БПГН или ТМА, а также рабдомиолиза при идиопатических воспалительных миопатиях, острой уратной нефропатии или острого тубулоинтерстициального нефрита, иногда развивающегося при синдроме Шегрена или IgG4-ассоциированной болезни. Дифференциальная диагностика различных причин прогрессирующего ухудшения функции почек часто невозможна без нефробиопсии, результаты которой имеют важное значение для выбора адекватной терапии.
Используемые источники
- Моисеев С.В., Буланов Н.М. Аутоиммунитет, аутовоспаление и почки. Клин фармакол тер 2022;31(4):7-17 [Moiseev S, Bulanov N. Autoimmunity, autoinflammation and kidneys. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(4):7-17 (In Russ.)].
- Levey AS, Eckardt KU, Dorman NM, et al. Nomenclature for kidney function and disease: report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int 2020;97(6):1117-29.
- Lameire NH, Levin A, Kellum JA, et al. Harmonizing acute and chronic kidney disease definition and classification: report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int 2021;100(3):516-26.
- Шилов Е.М., Козловская Н.Л., Коротчаева Ю.В. Клинические рекомендации по диагностике и лечению быстропрогрессирующего гломерулонефрита (экстракапиллярного гломерулонефрита с полулуниями). Нефрология 2015;19(6):73–82 [Shilov EM, Kozlovskaya NL, Korotchaeva JV. Clinical guidelines for diagnosis and treatment of rapidly progressive glomerulonephritis (extracapillary glomerulonephritis with crescent formation). Nephrology (SaintPetersburg) 2015;19(6):73-82 (In Russ.)].
- Jennette JC. Rapidly progressive crescentic glomerulonephritis. Kidney Int 2003;63:1164–77.
- Aydin Z, Turkmen K, Dede F, et al. Demographic, clinical and laboratory characteristics of rapidly progressive glomerulonephritis in Turkey: Turkish Society of Nephrology-Glomerular Diseases (TSN-GOLD) Working Group. Clin Exp Nephrol 2021;25(2):173-83.
- Буланов Н.М., Моисеев С.В., Новиков П.И. и др. Поражение почек при различных вариантах АНЦА-ассоциированных васкулитов. Клин фармакол тер 2016;25(5):23-9 [Bulanov NM, Moiseev SV, Novikov PI, et al. Renal involvement in ANCA-associated vasculitis. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2016;25(5):23-9 (In Russ.)].
- Щеголева Е.М., Буланов Н.М., Виноградова Е.С. и др. Варианты течения и исходы микроскопического полиангиита. Клин фармакол тер 2018;27(3):35-40 [Shchegoleva EM, Bulanov NM, Vinogradova ES, et al. Clinical variants and outcomes of microscopic polyangiitis. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2014;1:32-6 (In Russ.)].
- McAdoo SP, Pusey CD. Antiglomerular basement membrane disease. Semin Respir Crit Care Med 2018;39(4):494-503.
- Huart A, Josse AG, Chauveau D, et al; French Society of Hemapheresis. Outcomes of patients with Goodpasture syndrome: A nationwide cohort-based study from the French Society of Hemapheresis. J Autoimmun 2016;73:24-9.
- van Daalen EE, Jennette JC, McAdoo SP, et al. Predicting outcome in patients with anti-GBM glomerulonephritis. Clin J Am Soc Nephrol 2018;13(1):63-72.
- Moiseev S, Cohen Tervaert JW, Arimura Y, et al. 2020 international consensus on ANCA testing beyond systemic vasculitis. Autoimmun Rev 2020;19(9):102618.
- McAdoo SP, Tanna A, Hruskova Z, et al. Patients double-seropositive for ANCA and anti-GBM antibodies have varied renal survival, frequency of relapse, and outcomes compared to single-seropositive patients. Kidney Int 2017;92:693–702.
- Kidney Disease: Improving Global Outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical practice guideline for the management of glomerular diseases. Kidney Int 2021;100(4S):S1-S276.
- Fanouriakis A, Kostopoulou M, Cheema K, et al. 2019 Update of the Joint European League Against Rheumatism and European Renal AssociationEuropean Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of lupus nephritis. Ann Rheum Dis 2020;79(6):713-23.
- Буланов Н.М., Козловская Н.Л., Тао Е.А. и др. Современные подходы к лечению АНЦА-ассоциированных васкулитов с поражением почек с позиций медицины, основанной на доказательствах. Клин фармакол тер 2020;29(4):72-84 [Bulanov N, Kozlovskaya N, Tao E, et al. Evidence-based treatment of ANCA-associated vasculitis with kidney involvement. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2020;29(4):72-84 (In Russ.)].
- Jayne D, Walsh M, Merkel PA, et al. Plasma exchange and glucocorticoids to delay death or end-stage renal disease in anti-neutrophil cytoplasm antibody-associated vasculitis: PEXIVAS non-inferiority factorial RCT. Health Technol Assess 2022;26(38):1-60.
- Furie R, Rovin BH, Houssiau F, et al. Two-year, randomized, controlled trial of belimumab in lupus nephritis. N Engl J Med 2020;383(12):1117-28.
- Zhu Y, Rao J, Liu L, et al. The therapeutic effect of plasma exchange on ANCAassociated vasculitis: A meta-analysis. Clin Nephrol 2021;95(6):312-22.
- Bellos I, Michelakis I, Nikolopoulos D. The role of plasma exchange in antineutrophil cytoplasmic antibody-associated vasculitis: a meta-analysis. Clin Rheumatol 2021;40(4):1447-56.
- Козловская Н.Л., Прокопенко Е.И., Эмирова Х.М., Серикова С.Ю. Клинические рекомендации по диагностике и лечению атипичного гемолитикоуремического синдрома. Нефрология и диализ 2015;17(3):242-64 [Kozlovskaya NL, Prokopenko EI, Emirova KhM, Serikova SYu. Clinical guidelines for diagnosis and treatment of atypical hemolytic uremic syndrome. Nephrology and Dialysis 2015;17(3):242-64 (In Russ.)].
- Коротчаева Ю.В., Козловская Н.Л., Демьянова К.А. и др. Атипичный гемолитико-уремический синдром: клиническая картина, диагностика и лечение. Клин фармакол тер 2022;31(2):43-50 [Korotchaeva Yu, Kozlovskaya N, Demyanova K, et al. Atypical hemolytic-uremic syndrome: clinical presentation, diagnosis and treatment. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(2):43-50 (In Russ.)].
- Scheen M, Adedjouma A, Esteve E, et al. Kidney disease in antiphospholipid antibody syndrome: Risk factors, pathophysiology and management. Autoimmun Rev 2022;21(5):103072.
- Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 2017;91(3):539-51.
- Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;4(2):295-306.
- Сervera R, Piette JC, Font J, et al. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002;46(4):1019-27.
- Cervera R, Serrano R, Pons-Estel GJ, et al; Euro-Phospholipid Project Group (European Forum on Antiphospholipid Antibodies). Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis 2015;74(6):1011-8.
- Rodríguez-Pintó I, Moitinho M, Santacreu I, et al; CAPS Registry Project Group (European Forum on Antiphospholipid Antibodies). Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of 500 patients from the International CAPS Registry. Autoimmun Rev 2016;15(12):1120-4.
- Domingues V, Chock EY, Dufrost V, et al. Increased risk of acute and chronic microvascular renal lesions associated with antiphospholipid antibodies in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Autoimmun Rev 2022;21(10):103158.
- El Hasbani G, Viola M, Sciascia S, et al. Antiphospholipid antibodies in inflammatory and autoimmune rheumatic and musculoskeletal diseases beyond lupus: a systematic review of the available evidence. Rheumatol Ther 2021;8(1):81-94.
- Tektonidou MG, Andreoli L, Limper M, et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis 2019;78(10):1296-304.
- López-Benjume B, Rodríguez-Pintó I, Amigo MC, et al; on behalf the CAPS Registry Project Group/European Forum on Antiphospholipid Antibodies. Eculizumab use in catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis from the "CAPS Registry". Autoimmun Rev 2022;21(4):103055.
- Woodworth TG, Suliman YA, Li W, et al. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol 2016;12(11):678-91.
- Гордеев А.В., Захарова А.Ю., Мутовина З.Ю., Ананьева Л.П. Современные представления о гетерогенности поражений почек у больных склеродермией. Научно-практическая ревматология 2015;53(4):343-445 [Gordeev AV, Zakharova AYu, Mutovina ZYu, Ananyeva LP. Current views on the heterogeneity of renal involvement in patients with systemic sclerosis. Rheumatology Science and Practice 2015;53(4):343-445 (In Russ.)].
- Cole A, Ong VH, Denton CP. Renal disease and systemic sclerosis: an update on scleroderma renal crisis. Clin Rev Allergy Immunol 2022 Jun 1. PMID: 35648373.
- Turk M, Pope JE. The frequency of scleroderma renal crisis over time: a metaanalysis. J Rheumatol 2016;43(7):1350–1355.
- Bütikofer L, Varisco PA, Distler O, et al; EUSTAR collaborators. ACE inhibitors in SSc patients display a risk factor for scleroderma renal crisis - a EUSTAR analysis. Arthritis Res Ther. 2020;22(1):59.
- Pestaña-Fernández M, Rubio-Rivas M, Tolosa-Vilella C, et al; for RESCLE Investigators, Autoimmune Diseases Study Group (GEAS). The incidence rate of pulmonary arterial hypertension and scleroderma renal crisis in systemic sclerosis patients with digital ulcers on endothelin antagonist receptors (ERAs) and phosphodiesterase-5 inhibitors (PDE5i). Rheumatology (Oxford) 2021;60(2):872-80.
- Butler EA, Baron M, Fogo AB, et al; Scleroderma Clinical Trials Consortium Scleroderma Renal Crisis Working Group. Generation of a сore set of items to develop classification criteria for scleroderma renal crisis using consensus methodology. Arthritis Rheumatol 2019;71(6):964-71.
- Yamashita H, Kamei R, Kaneko H. Classifications of scleroderma renal crisis and reconsideration of its pathophysiology. Rheumatology (Oxford) 2019;58:2099–106.
- Moinzadeh P, Kuhr K, Siegert E, et al. Scleroderma renal crisis: risk factors for an increasingly rare organ complication. J Rheumatol 2020;47(2):241–8.
- Kim H, Lefebvre F, Hoa S, Hudson M. Mortality and morbidity in scleroderma renal crisis: A systematic literature review J Scleroderma Relat Disord 2021; 6(1):21-36.
- Kowal-Bielecka O, Fransen J, Avouac J, et al; EUSTAR Coauthors. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2017;76(8):1327-39.
- Stern EP, Host LV, Wanjiku I, et al. Zibotentan in systemic sclerosis-associated chronic kidney disease: a phase II randomised placebo-controlled trial. Arthritis Res Ther 2022;24(1):130.
- Hoste EA, Bagshaw SM, Bellomo R, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med 2015;41(8):1411-23.
- Захарова Е.В., Остроумова О.Д., Клепикова М.В. Лекарственно-индуцированное острое повреждение почек. Безопасность и риск фармакотерапии. 2021;9(3):117–27 [Zakharova EV, Ostroumova OD, Klepikova MV. Druginduced acute kidney injury. Bezopasnost’ i risk farmakoterapii = Safety and Risk of Pharmacotherapy 2021;9(3):117–127 (In Russ.)].
- Perazella MA, Rosner MH. Drug-induced acute kidney injury. Clin J Am Soc Nephrol 2022;17(8):1220-33.
- Kellum JA, Romagnani P, Ashuntantang G, et al. Acute kidney injury. Nat Rev Dis Primers 2021;7(1):52.
- Perazella MA, Markowitz GS. Bisphosphonate nephrotoxicity. Kidney Int 2008;74(11):1385-93.
- Toussaint ND, Elder GJ, Kerr PG. Bisphosphonates in chronic kidney disease; balancing potential benefits and adverse effects on bone and soft tissue. Clin J Am Soc Nephrol 2009;4(1):221-33.
- Bentata Y. Tacrolimus: 20 years of use in adult kidney transplantation. What we should know about its nephrotoxicity. Artif Organs 2020;44(2):140-152.
- Caravaca-FontЗn F, FernЗndez-JuЗrez G, Praga M. Acute kidney injury in interstitial nephritis. Curr Opin Crit Care 2019;25(6):558-564.
- Cucchiari D, Angelini C. Renal involvement in idiopathic inflammatory myopathies. Clin Rev Allergy Immunol 2017;52(1):99-107.
- Couvrat-Desvergnes G, Masseau A, Benveniste O, et al. The spectrum of renal involvement in patients with inflammatory myopathies. Medicine (Baltimore) 2014;93(1):33-41.
- Jasiek M, Karras A, Le Guern V, et al. A multicentre study of 95 biopsy-proven cases of renal disease in primary Sjögren's syndrome. Rheumatology (Oxford) 2017;56(3):362-70.
- Kawano M, Saeki T, Ubara Y, Matsui S. Recent advances in IgG4-related kidney disease. Mod Rheumatol 2022 Jul 5:roac065.
- Calatroni M, Moroni G, Reggiani F, Ponticelli C. Renal sarcoidosis. J Nephrol 2022 Jun 27.
- Muriithi AK, Leung N, Valeri AM, et al. Biopsy-proven acute interstitial nephritis, 1993-2011: a case series. Am J Kidney Dis 2014;64(4):558-66.
- Nakaosa N, Tsuboi N, Okabayashi Y, et al. Tubulointerstitial nephritis: a biopsy case series of 139 Japanese patients. Clin Exp Nephrol 2022;26(5):435-44.
- Фомин В.В. Подагрическая (уратная) нефропатия. Consilium Medicum 2021;23(1):11–4 [Fomin VV. Gouty (urate) nephropathy. Consilium Medicum 2021;23(1):11–4 (In Russ.)].