Оценка фармакокинетики, безопасности и переносимости препарата Ранквилон® у здоровых добровольцев: результаты клинического исследования I фазы
Сравнительная оценка фармакокинетики, безопасности и переносимости препарата Ранквилон® в таблетках 1 мг при однократном применении натощак и после приема пищи у здоровых добровольцев мужского и женского пола.
Проведено открытое проспективное рандомизированное трехпериодное, перекрестное исследование сравнительной фармакокинетики препарата Ранквилон® в таблетках 1 мг с тремя последовательными приемами однократной дозы 3 мг (3 таблетки по 1 мг) натощак и после приема пищи у 26 здоровых добровольцев (мужчин и женщин). В каждом периоде исследования отбирали 16 проб крови: непосредственно перед и в течение 10 ч после приема исследуемого препарата. Количественное определение действующего вещества в образцах плазмы крови проводили методом высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием. Значение влияния приема пищи на фармакокинетику препарата Ранквилон® определяли на основании критериев биоэквивалентности показателей AUC(0-t) и Cmax до и после приема пищи. Оценивали также безопасность и переносимость препарата Ранквилон®.
Прием пищи оказывал существенное влияние на AUC(0-t) исследуемого препарата, так как отношение средних геометрических значений этого показателя натощак и после приема пищи составило 81,75% (90% доверительный интервал [ДИ] 74,25–90,01%), что выходит за пределы 90% ДИ 80,00–125,00%. В то же время прием пищи существенно не влиял на Cmax препарата: отношение средних геометрических значений этого показателя натощак и после приема пищи составило 89,77% (90% ДИ: 81,82–98,5%), т.е. укладывалось в границы биоэквивалентности 80,00–125,00%. Средние значения AUC(0-t) после однократного приема здоровыми добровольцами препарата Ранквилон® натощак составили 17,502±8,587 нг×ч/мл, а после приема пищи – 21,971 ±13,015 нг×ч/мл. Таким образом, при приеме препарата Ранквилон® после приема пищи показатель AUC(0-t) увеличивается примерно в 1,26 раза по сравнению с приемом препарата натощак. Во время исследования было зарегистрировано одно нежелательное явление у одного добровольца (головная боль).
С учетом биодоступности и профиля безопасности препарат Ранквилон® в таблетких 1 мг предпочтительно принимать во время или после приема пищи.
Тревожные расстройства относятся к одним из наиболее распространенных в популяции психических нарушений и являются важной медицинской, социальной и экономической проблемой. Согласно статистическим данным, до 20% взрослого населения страдает различными расстройствами тревожного спектра [1–3].
Значимую роль в структуре нозологий, сопровождающихся тревогой, играют нарушения адаптации и неврастения. Распространенность диагностированного расстройства адаптации достаточно высока и достигает 10% в общей медицинской и 25% в амбулаторной психиатрической практике. Расстройство адаптации представляет собой состояние субъективного дистресса, сопровождающееся психопатологическими нарушениями с трудностями в социальной и трудовой деятельности, обычно возникающее в периоды значительных изменений в жизни или во время стрессовых событий. При этом самым распространенным вариантом (17– 29% случаев) является расстройство адаптации с тревожной симптоматикой. Наличие данных расстройств повышает чувствительность пациентов к стрессовым нагрузкам, предрасполагает к формированию депрессии, алкоголизма, декомпенсации личностных расстройств и риску суицида [4].
Распространенность неврастении среди пациентов общей медицинской практики в среднем составляет 1,3–5,2% [5]. Снижая физические и интеллектуальные способности человека, неврастения негативно влияет на качество жизни [6]. Наряду с повышенной утомляемостью, вялостью, слабостью, снижением активности важное место в клинической картине неврастении занимают так называемые гиперестетические расстройства, такие как тревожность, раздражительность, эмоциональная лабильность, что обосновывает применение для терапии таких состояний препаратов с анксио литическим действием. Однако существующие анксиолитические препараты обладают недостаточной эффективностью и вызывают побочные эффекты, а зачастую и углубление астенический симптоматики.
Таким образом, проблема терапии тревожных расстройств является одной из наиболее важных в психиатрии и психофармакологии. Препарат Ранквилон® представляет собой оригинальное анксиолитическое лекарственное средство пептидной структуры, которое предназначено для применения у взрослых пациентов с тревожными состояниями при неврастении и расстройствах адаптации. Препарат представляет собой антагонист центральных холецистокининовых рецепторов, что подтверждается совокупными данными радиолигандного анализа и исследованиями функционального антагонизма с тетрапептидом холецистокинина [7]. Исходя из важного значения холецистокининовой системы в этиологии и патогенезе тревожных расстройств, можно полагать, что Ранквилон® будет эффективным в лечении состояний, сопровождающихся тревогой. Следует отметить, что у данного препарата отсутствуют разрешенные к медицинскому применению аналоги по механизму действия. Появление нового небензодиазепинового анксиолитика позволит повысить доступность и эффективность терапии у пациентов с тревожными расстройствами.
В соответствии с руководствами по проведению доклинических и клинических исследований лекарственных средств ФГБУ "Научный центр средств медицинского применения" Минздрава России, а также Решением Коллегии Евразийской экономической комиссии от 26.11.2019 г. № 202 "Об утверждении Руководства по доклиническим исследованиям безопасности в целях проведения клинических исследований и регистрации лекарственных препаратов" и Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 78 "О Правилах регистрации и экспертизы лекарственных средств для медицинского применения" [8-11] при разработке препарата Ранквилон® были проведены фармакологические, фармакокинетические и токсикологические доклинические исследования и клинические исследования I, II и III фаз. В настоящей статье приведены результаты исследования влияния приема пищи на фармакокинетику, безопасность и переносимость препарата Ранквилон® у здоровых добровольцев [11].
Цель исследования – сравнительная оценка фармакокинетики, безопасности и переносимости препарата Ранквилон® в таблетках 1 мг при однократном применении натощак и после приема пищи у здоровых добровольцев мужского и женского пола.
Материал и методы
ное, перекрестное, сравнительное исследование фармакокинетики препарата Ранквилон® в таблетках 1 мг (АО "Валента Фарм", Россия) с тремя последовательными приемами однократной дозы натощак (fasting) и после приема пищи (fed) у здоровых мужчин и женщин. Добровольцы были рандомизированы на две группы в зависимости от последовательности приема препарата: 1-я (n=13) – натощак/натощак/после приема пищи, 2-я (n=13) – после приема пищи/после приема пищи/натощак.
Задачами настоящего исследования были определение концентрации амида N-(6-фенилгексаноил)глицил-L-триптофана в плазме крови добровольцев после однократного применения препарата натощак или после приема пищи в каждом из трех периодов исследования; оценка фармакокинетических параметров и относительной биодоступности препарата Ранквилон® в каждом из трех периодов исследования; сравнение фармакокинетических параметров и относительной биодоступности препарата Ранквилон® при применении натощак и после приема пищи; оценка профиля безопасности препарата Ранквилон® в каждом из трех периодов исследования (частота возникновения нежелательных явлений [НЯ] и серьезных нежелательных явлений [СНЯ], изменения данных лабораторных исследований, физического осмотра, функций жизненно важных органов); оценка переносимости препарата Ранквилон® (доля добровольцев, которые досрочно прекратили участие в исследовании из-за НЯ/СНЯ с оценкой времени до выбывания).
Исследование было начато после получения Разрешения на проведение клинического исследования Министерства здравоохранения РФ (№ 623 от 11 октября 2021 г.), одобрения Совета по Этике при Министерстве здравоохранения РФ (выписка из протокола № 287 от 14 сентября 2021 г.) и одобрения независимого (локального) этического комитета исследовательского центра (выписка из протокола № 179 Этического комитета "БиоЭтика" от 3 ноября 2021 г.).
Участники исследования были отобраны согласно следующим критериям включения: подписанное информированное согласие на участие в исследовании; мужчины и женщины в возрасте 18–45 лет с индексом массы тела (ИМТ) от 18,5 до 30 кг/м2, масса тела у мужчин и женщин >55 кг и >45 кг, соответственно; отсутствие каких-либо острых или хронических заболеваний; отрицательные результаты тестов на ВИЧ, сифилис, гепатит В и С; отказ от приема алкоголя за 2 недели до первого приема препарата и в течение всего исследования; отрицательные тесты на содержание паров алкоголя в выдыхаемом воздухе и содержание наркотических веществ в моче; отрицательный тест на беременность у женщин с сохраненным репродуктивным потенциалом; согласие придерживаться эффективных методов контрацепции на протяжении всего исследования и одного месяца после его завершения.
Критериями невключения были невозможность получения информированного согласия; хирургические вмешательства на желудочно-кишечном тракте в анамнезе (за исключением аппендэктомии); систолическое артериальное давление <100 мм рт. ст. или >130 мм рт. ст.; диастолическое артериальное давление <60 мм рт. ст. или >90 мм рт. ст.; частота сердечных сокращений <60 в минуту или >90 в минуту, температура тела <35,5оС или >36,9оС, частота дыхания <12 или >20 в минуту; наличие в анамнезе острых или хронических заболеваний; активные инфекции, включая локальные, менее чем за 4 недели до скрининга; непереносимость лактозы, дефицит лактазы или глюкозогалактозная мальабсорбция; повышенная чувствительность к любому компоненту исследуемого лекарственного препарата, к гепарину или гепарин-индуцированная тромбоцитопения в анамнезе; отягощенный аллергологический анамнез; прием более 5 единиц алкоголя в неделю в анамнезе и/или анамнестические сведения об алкоголизме, наркомании, злоупотреблении лекарственными препаратами; прием любых лекарственных препаратов, включая витамины, растительные препараты, а также биологически активных добавок менее чем за 30 дней до скрининга; применение гормональных контрацептивов добровольцами женского пола менее чем за 2 мес до скрининга; курение более 10 сигарет в сутки; отклонение лабораторных показателей от референсных значений лаборатории, клинически значимые отклонения на электрокардиограмме (ЭКГ); донорство (450 мл крови и более) менее чем за 2 мес до скрининга; соблюдение любой диеты с низким содержанием натрия либо соблюдение поста и особой диеты, ведение особого образа жизни, которые могут препятствовать проведению исследования; беременность, период грудного вскармливания или планирование беременности в период участия в исследовании и в течение одного месяца после его завершения; участие в любом клиническом исследовании лекарственного препарата или терапевтического устройства в течение 3 мес или как минимум 5 периодов полувыведения препарата в зависимости от того, что дольше, до скрининга.
Схема исследования приведена на рис. 1. Исследование состояло из периода скрининга, периодов исследования фармакокинетики препарата Ранквилон® – I, II и III периоды, соответственно, разделенных I и II периодами "отмывки". Общая продолжительность клинического исследования для одного добровольца (с учетом периода скрининга) составляла не более 30 сут:
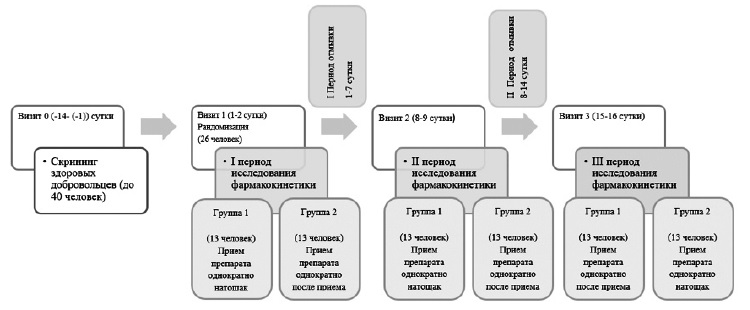
В каждом периоде исследования добровольцы принимали препарат Ранквилон® утром натощак или после приема пищи однократно в дозе 3 мг. В случае применения препарата после приема пищи добровольцы начинали ее прием за 30 минут до приема препарата. Завтрак был высококалорийным (800–1000 ккал) с высоким содержанием жиров (около 50% от общей калорийности). Таблетки проглатывали целиком, не разжевывая и запивая 200 мл воды.
В каждом периоде исследования фармакокинетики производили отбор 16 проб крови: непосредственно перед приемом исследуемого препарата (00:00) (проба 0) и через 00:15, 00:30, 00:45, 01:00, 01:15, 01:30, 01:45, 02:00, 02:30, 03:00, 04:00, 05:00, 06:00, 08:00, 10:00 часов:минут после приема препарата. Образцы крови объемом 6 мл забирали в пробирки (вакутейнеры) с антикоагулянтом (К2 ЭДТА). До центрифугирования образцы крови хранили на ледяной бане (от 0 до +2оС). Образцы крови центрифугировали не позднее, чем через 30 мин после взятия крови в течение 10 мин при 3000 g и температуре +4±2оС. Все образцы полученной плазмы крови делили на две аликвоты. До отправки в лабораторию все образцы плазмы хранили в морозильной камере в исследовательском центре при контролируемой температуре -70оС (±10оС).
Количественное определение амида N-(6-фенилгексаноил)глицил-L-триптофана в образцах плазмы крови проводили валидированным методом высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием (ВЭЖХ-МС/МС). Нижний предел количественного определения (НПКО) амида N-(6-фенилгексаноил)глицил-L-триптофана в плазме крови составил 0,2 нг/мл, линейный диапазон – 0,2–20 нг/мл. В качестве метода пробоподготовки использовали жидкость-жидкостную экстракцию метил-трет-бутиловым эфиром. Разработанная методика была валидирована по следующим параметрам: селективность, специфичность, эффект матрицы, калибровочная кривая (линейность), точность, прецизионность, степень извлечения, НПКО, предел обнаружения, перенос пробы, стабильность.
Основными фармакокинетическими параметрами были максимальное измеренное значение концентрации действующего вещества в плазме крови добровольца (Cmax) и суммарная площадь под фармакокинетической кривой, начиная с нулевого значения времени (момент приема препарата) до времени отбора последнего образца крови с концентрацией выше НПКО (AUC(0-t)).
Также анализировали дополнительные фармакокинетические параметры: время достижения максимальной концентрации действующего вещества в плазме крови добровольца (t|max), время от точки 0 до первой точки с концентрацией выше НПКО (tlag), площадь под фармакокинетической кривой "плазменная концентрация – время", экстраполированная до бесконечности (AUC(0-∞)); доля (в %) AUC(0-t) от AUC(0-∞); константа элиминации, которую рассчитывали по значениям фармакокинетического профиля на терминальном участке кривой с использованием не менее 3 значений (последние 3 значения); период полувыведения (t1/2); среднее время удержания препарата в крови (MRTlast); кажущийся объем распределения (Vd); клиренс (CL); относительная биодоступность, рассчитанная по формуле f’=AUC(0-t)(fasting)/AUC(0-t)(fed); относительная скорость всасывания, рассчитанная по формуле f" =Cmax(fasting)/Cmax(fed)
Оценка влияния приема пищи на фармакокинетику препарата Ранквилон® осуществлялась на основании критериев биоэквивалентности показателей AUC(0-t) и Cmax до и после приема пищи.
Безопасность исследуемого препарата оценивали по следующим параметрам:- НЯ и СНЯ (собирали всю информация о НЯ/СНЯ, а также сопутствующей терапии по поводу НЯ/СНЯ).
- Изменения данных рутинных лабораторных обследований (клинический анализ крови, общий анализ мочи, биохимический анализ крови), ЭКГ, данных физического исследования и жизненно-важных показателей.
Переносимость исследуемого препарата оценивали на основании доли добровольцев, которые досрочно прекратили участие в исследовании из-за НЯ/СНЯ, и времени до выбывания.
Статистические методы. Популяция для статистического анализа состояла из всех добровольцев, которые завершили три периода исследования фармакокинетики. Популяция безопасности в настоящем исследовании включала всех добровольцев, принявших исследуемый препарат хотя бы один раз.
Для анализа фармакокинетики использовали программу WinNonlin 8.0 (Pharsight® Corporation, Certara USA, Inc., Princeton, NJ, USA). Для описательной статистики исходных параметров и других зарегистрированных клинических данных была использована программа WinNonlin 8.0.
Результаты
Характеристика добровольцев. Из 36 здоровых добровольцев, прошедших скрининг, 26 были рандомизированы для участия в исследовании: 13 добровольцев (5 мужчин и 8 женщин, средний возраст 29±7 лет) получали препарат в последовательности натощак/ натощак/после приема пищи (1-я группа) и 13 добровольцев (6 мужчин и 7 женщин, средний возраст 28±5 лет) – после приема пищи/после приема пищи/натощак (2-я группа). Все добровольцы были представителями европеоидной расы. Средний ИМТ составил 23,1±2,4 кг/м2 в 1-й группе и 23,0±2,4 кг/м2 во 2-й группе.
Все добровольцы завершили исследование по протоколу и были включены в анализ фармакокинетики.
Результаты анализа фармакокинетики. Полученные усредненные фармакокинетические кривые зависимо- сти концентрации амида N-(6-фенилгексаноил)глицил- L-триптофана от времени представлены на рис. 2.
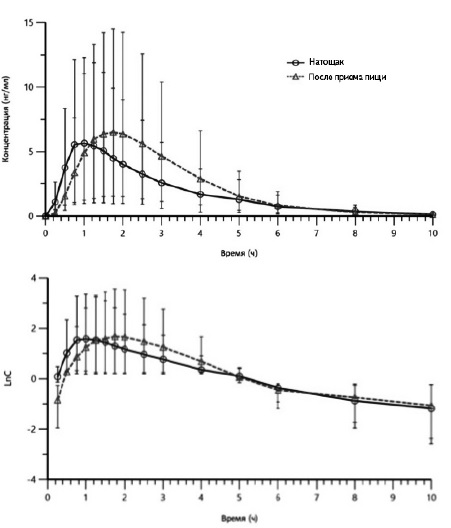
Средние значения фармакокинетических параметров амида N-(6-фенилгексаноил)глицил-L-триптофана после однократного применения препарата натощак или после приема пищи приведены в табл. 1.
| Параметр | Натощак | После приема пищи |
|---|---|---|
| Примечания: данные представлены как среднее ± стандартное отклонение; *данные представлены в виде медианы и диапазона (минимум – максимум). | ||
| Cmax (нг/мл) | 7,329±3,708 | 8,482±4,781 |
| AUC(0-t) (ч×нг/мл) | 17,502±8,587 | 21,971±13,015 |
| AUC(0-∞) (ч×нг/мл) | 18,448±8,653 | 22,682±13,229 |
| AUC%inf (%) | 94,037±7,011 | 96,414±2,216 |
| tlag (ч)* | 0 (0–0,25) | 0,25 (0–0,75) |
| tmax (ч)* | 1,0 (0,5–3,0) | 1,5 (0,75–4,0) |
| t1/2 (ч) | 1,811±0,902 | 1,396±0,582 |
| Kel (ч-1) | 0,455±0,179 | 0,561±0,179 |
| MRTlast (ч) | 2,564±0,622 | 2,645±0,694 |
| Vz (Vd) (л) | 173,891±143,708 | 111,427±72,881 |
| Cl (л/ч) | 67,885±35,375 | 59,548±34,37 |
Относительная биодоступность амида N-(6-фенилгексаноил)глицил-L-триптофана после однократного приема препарата натощак и после приема пищи составила: показатель степени абсорбции ln(AUC(0-t)) [fasting]/ ln(AUC(0-t)) [fed] (f’) – 88,042±42,328%, показатель скорости абсорбции ln(Cmax) [fasting]/ln(Cmax) [fed] – 95,571±38,272%.
Был проведен дисперсионный анализ, в модель которого следующие переменные включали в качестве фиксированных эффектов (согласно Правилам ЕАЭС): "Последовательность", "Период", "Лекарственный препарат" и случайный фактор "Субъект после дова тельности".
Влияние приема пищи на биодоступность препарата Ранквилон® оценивали на основании критериев био эквивалентности показателей AUC(0-t) и Cmax до и после приема пищи. Прием пищи оказывал статистически значимое влияние на AUC(0-t), так как относительная биодоступность препарата Ранквилон® выходит за пре делы 90% доверительного интервала (ДИ) 80,00– 125,00%. Групповые оценки отношений среднего значения относительной биодоступности препарата Ранквилон® f’ = AUC(0-t) [fasting]/AUC1(0-t) [fed] (GeoLSM) составили 81,75% (90% ДИ 74,25–90,01%).
В то же время влияние приема пищи на Cmax препарата оказалось несущественным, так как относительная биодоступность препарата Ранквилон® не выходила за пределы 90% ДИ 80,00–125,00%. Групповые оценки отношений среднего значения f" = Cmax[fasting]/Cmax [fed] (GeoLSM) составили 89,77% (90% ДИ 81,82– 98,5%).
По результатам проведенного анализа мы не выявили статистически значимого вклада факторов "Прием пищи" и "Последовательность" на дисперсию логарифмически преобразованных фармакокинетических данных lnCmax и фактора "Последовательность" на дисперсию логарифмически преобразованных фармакокинетических данных lnAUC(0-t). Основными факторами, вносящими значимый вклад в наблюдаемую вариабельность данных, являются "Испытуемые" (p<0,0001 для lnAUC(0-t) и lnCmax p<0,0001), "Этап (период) исследования" (р=0,0077 для lnCmax) и "Прием пищи" (р=0,001 для lnAUC(0-t)).
Среднее значение AUC(0-t) после однократного приема здоровыми добровольцами препарата Ранквилон® натощак составило 17,502±8,587 нг×ч/мл, после приема пищи – 21,971±13,015 нг×ч/мл. Таким образом, при приеме препарата Ранквилон® после приема пищи AUC(0-t) увеличивается примерно в 1,26 раза по сравнению с приемом препарата натощак.
Безопасность и переносимость. В анализ безопасности и переносимости исследуемого препарата включили данные всех 26 рандомизированных добровольцев. Все добровольцы приняли исследуемый препарат согласно протоколу в целом 9 мг за все исследование (3 таблетки по 1 мг в каждом периоде). Во время исследования зафиксировано одно НЯ (головная боль) у одного добровольца. НЯ было легкой степени тяжести, связь с исследуемым препаратом – возможная, завершилось выздоровлением и не потребовало медицинского вмешательства. НЯ наблюдалось после применения исследуемого препарата после приема пищи. Таким образом, общая частота НЯ в данном исследовании составила 3,8% (1/26).
Клинически значимых отклонений жизненно важных показателей и результатов физического обследования в ходе исследования не зарегистрировали. Все параметры ЭКГ и показатели лабораторных исследований (клинический анализ крови, общий анализ мочи, биохимический анализ крови) были в пределах нормальных референсных значений при проведении анализов на скрининге и при завершении исследования.
Доля добровольцев, которые досрочно прекратили участие в исследовании из-за возникновения НЯ/СНЯ, была равна 0. Единственное зарегистрированное в ходе исследования НЯ (головная боль) не соответствовало критериям досрочного выбывания добровольца из исследования.
Обсуждение
Прием пищи является одним из наиболее значимых факторов, оказывающих влияние на биодоступность лекарственных средств. На этапе всасывания лекарственного препарата в желудочно-кишечном тракте пища может изменять скорость растворения действующего вещества, значение рН в желудке и двенадцатиперстной кишке, удельную площадь всасывания лекарственного средства за счет перераспределения химуса в просвете кишечника и другие физиологические параметры. Лекарственный препарат может также конкурировать с компонентами пищи за белки-переносчики и мембранные рецепторы, которые влияют на абсорбцию препарата. Помимо этого, содержащиеся в пище молекулы белков, жиров и углеводов могут образовывать комплексы с лекарственным средством, что также может изменять фармакокинетические и фармакодинамические параметры препарата. На этапе метаболизма в печени пищевые продукты могут как индуцировать, так и ингибировать ферменты системы цитохрома Р450 и Р-гликопротеина [12]. Вышеописанные взаимодействия могут значительным образом изменять биодоступность вводимого препарата, что следует учитывать при разработке новых препаратов и особенно их пероральных лекарственных форм.
Фармацевтической компанией АО "Валента Фарм" в сотрудничестве с ФГБНУ "НИИ фармакологии имени В.В. Закусова" был разработан новый оригинальный препарат Ранквилон® в лекарственной форме таблетки по 1 мг. Проведенное исследование позволило установить, что прием препарата вместе с пищей в 1,26 раза увеличивает значение AUC(0-t) по сравнению с приемом препарата натощак.
Заключение
В проведенном исследовании изучались фармакокинетические параметры препарата Ранквилон® в форме таблеток по 1 мг в дозе 3 мг при однократном применении натощак и после приема пищи. Было установлено, что прием пищи статистически значимо повышает экспозицию препарата (значение AUC(0-t)), но не оказывает значимого влияния на показатель Cmax. Препарат показал хороший профиль безопасности – на протяжении всего исследования было зарегистрировано только одно НЯ легкой степени тяжести (головная боль) в группе добровольцев, принимавших препарат после приема пищи. Результаты проведенного исследования позволяют сделать вывод о том, что с учетом биодоступности и профиля безопасности препарат Ранквилон® в таблетках 1 мг предпочтительно принимать во время или после приема пищи.
Используемые источники
- Munir S, Takov V. Generalized anxiety disorder. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 [cited 2022 Dec 24]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK441870/
- Сиволап Ю.П. Систематика и лечение тревожных расстройств. Журнал неврологии и психиатрии им. С.С. Корсакова 2020;120(7):121-7 [Sivolap YuP. Systematics and treatment of anxiety disorders. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova. 2020;120(7):121-7. (In Russ.)].
- Bandelow B, Michaelis S. Epidemiology of anxiety disorders in the 21st century. Dialogues Clin Neurosci 2015 Sep;17(3):327–35.
- Вельтищев Д. Ю. Расстройство адаптации как стрессовый синдром: психопатология и терапия. Доктор.Ру 2013;5(83):76–81 [Veltishchev DYu. Adjustment disorder as stress syndrome: psychopathology and treatment. Doctor.Ru 2013;5(83):76–81 (In Russ.)].
- Добрушина О.Р., Медведев В.Э. Сочетанная терапия неврастении в общей медицинской практике. Consilium Medicum 2016;18(2):95–9 [Dobrushina OR, Medvedev VE. Combined therapy of neurasthenia in general medical practice. Consilium Medicum 2016;18(2):95–9 (in Russ.)].
- Дадашева К.Н., Агафонов Б.В., Дадашева М.Н., Подрезова Л.А. Неврастенический синдром в общеврачебной практике. Возможности терапии. РМЖ. Медицинское обозрение 2019;3(4(II)):91-5 [Dadasheva KN, Agafonov BV, Dadasheva MN, Podrezova LA. Asthenic syndrome in general practice: therapy possibilities. RMJ. Medical Review 2019;4(II):91–5 (in Russ.)].
- Колик Л.Г., Гудашева Т.А., Середенин С.Б. Об участии холецистокининовой системы в реализации анксиолитических эффектов дипептида ГБ-115. Бюллень экспериментальной биологии и медицины 2012;153(6):828-32 [Kolik LG, Gudasheva TA, Seredenin SB. Role of the cholecystokinin system in anxiolytic activity of dipeptide GB-115. Bullen Experimental Biology and Medicine 2012;153(6):828-32 (In Russ.)].
- Решение Коллегии Евразийской экономической комиссии от 26.11.2019 г. № 202 “Об утверждении Руководства по доклиническим исследованиям безопасности в целях проведения клинических исследований и регистрации лекарственных препаратов”.
- Решение Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 78 “О Правилах регистрации и экспертизы лекарственных средств для медицинского применения”. Решение Совета Евразийской экономической комиссии от 3 ноября 2016 г. № 78 “О Правилах регистрации и экспертизы лекарственных средств для медицинского применения”.
- Руководство по проведению доклинических исследований лекарственных средств. Миронов А.Н. Том 1. Гриф и К. 2012.
- Отчет о клиническом исследовании № РАН-01-02-2021 “Открытое клиническое исследование I фазы по изучению влияния приема пищи на фармакокинетику препарата Ранквилон®, таблетки, 1 мг, у здоровых добровольцев при однократном применении” 2021.
- Кирилюк А.А., Петрище Т.Л. Особенности влияния пищевых продуктов и их компонентов на фармакологическую активность лекарственных средств. Современные проблемы здравоохранения и медицинской статистики 2017;1:51-63 [Kirilyuk AA, Petrishche TL. Characteristics of the influence of food products and their components for pharmacological activity of drugs. Current problems of health care and medical statistics 2017;1:51-63 (In Russ.)].