Редкие (орфанные) наследственные заболевания с поражением почек: подходы к диагностике и лечению
Возможные варианты поражения почек при редких наследственных заболеваниях включают в себя поражение клубочков и канальцев, аномалии почек и мочевыводящих путей, уролитиаз, множественные кисты, злокачественные или доброкачественные опухоли. Предполагать наследственный генез нефропатии следует в первую очередь в случае ее развития в детском, подростковом или молодом возрасте и/или при наличии семейного анамнеза, хотя некоторые наследственные заболевания проявляются в старшем и даже пожилом возрасте, а другие случаи болезни среди членов семьи пробанда могут отсутствовать. При некоторых заболеваниях важное диагностическое значение имеют внепочечные проявления (например, нейросенсорная тугоухость при синдроме Альпорта, вихревидная кератопатия и ангиокератомы при болезни Фабри, светобоязнь при нефропатическом цистинозе). Для подтверждения диагноза моногенного наследственного заболевания необходимо молекулярно-генетическое исследование. В последние годы с этой целью стали шире использовать полноэкзомное секвенирование, прежде всего в тех случаях, когда возможной причиной поражения почек могут быть различные наследственные болезни со сходным фенотипом.
Редкие, или орфанные (от греч. cирота), заболевания по определению встречаются у небольшой части популяции. Однако сегодня известно около 7000 редких болезней, а общее количество таких пациентов в мире достигает 260-450 млн человек [1,2]. Критерии выделения редких болезней отличаются в разных странах. В США и Европейском Союзе редкими считают заболевания, распространенность которых в общей популяции ниже 1 на 1500 и 2000 населения, соответственно, а в Российской Федерации – 1 на 10000. Следует учитывать, что распространенность некоторых генетических заболеваний зависит от географического региона. Так, периодическая болезнь (семейная средиземноморская лихорадка) в нашей стране встречается редко (в основном у армян), в то время как в Турции количество таких пациентов исчисляется десятками тысяч [3]. Около 80% орфанных заболеваний являются генетически обусловленными (наследственными). Поражение почек отмечается примерно при 200 орфанных заболеваниях [1], а наследственными болезнями почек страдают около 10% взрослых и практически все дети, нуждающиеся в заместительной почечной терапии [4].
В последние годы возможности диагностики наследственных орфанных заболеваний значительно расширились за счет внедрения в клиническую практику и постепенного снижения стоимости генетических тестов. Одновременно увеличивается и количество орфанных препаратов, предназначенных для их лечения, что отражает меры по стимулированию разработки таких лекарственных средств, которые предпринимаются правительствами экономически развитых стран и включают в себя налоговые льготы, упрощенную процедуру допуска, государственное субсидирование разработки, увеличение срока эксклюзивности на рынке и др. [5]. Дос туп ность патогенетического лечения повышает актуальность своевременной диагностики соответствующих орфанных заболеваний с целью эффективной профилактики их прогрессирования и развития неблагоприятных исходов.
Подходы к диагностике
Наследственные заболевания обычно проявляются в детском или подростковом возрасте, однако генетические нефропатии не следует считать исключительно педиатрической проблемой. Во-первых, выживаемость пациентов с редкими заболеваниями почек увеличилась в результате разработки методов патогенетической терапии некоторых наследственных болезней и/или улучшения качества медицинской помощи в целом (прежде всего за счет диализа и трансплантации почки). Соответственно, после достижения 18-летнего возраста такие пациенты переходят под наблюдение нефрологов или других врачей, оказывающих помощь взрослым людям. Во-вторых, информированность врачей о редких заболеваниях остается низкой, поэтому диагноз нередко устанавливают спустя годы или десятилетия после появления первых симптомов. В-третьих, при некоторых наследственных заболеваниях первые симптомы могут появиться в старшем или даже пожилом возрасте. Соответственно, позднее развитие нефропатии не исключает ее наследственный генез.
Диагностика редких наследственных заболеваний с поражением почек производится в несколько этапов: (1) анализ семейного анамнеза; (2) определение фенотипа нефропатии; (3) выявление внепочечных проявлений; (4) молекулярно-генетический тест и/или другие исследования, необходимые для подтверждения диагноза [1].
Семейный анамнез. Изучение семейного анамнеза позволяет, хотя и не всегда, не только заподозрить и подтвердить наследственный характер поражения почек, но и выявить другие случаи заболевания, в том числе на более ранних стадиях, у родственников пробанда [6]. При некоторых наследственных заболеваниях семейный скрининг является наиболее эффективным методом диагностики. Например, в российской популяции у 165 (66,5%) из 248 пациентов с болезнью Фабри диагноз был установлен в результате обследования родственников пробандов, причем патогенные мутации гена GLA были выявлены примерно у половины потенциальных их носителей [7]. Однако семейный анамнез может отсутствовать, прежде всего при заболеваниях, которые передаются по аутосомно-рецессивному типу. При построении генеалогического дерева необходимо учитывать и возможную вариабельность фенотипов системных наследственных заболеваний, характеризующихся разнообразными клиническими проявлениями, у членов семьи пробанда.
Фенотип поражения почек. Возможные варианты поражения почек при наследственных орфанных заболеваниях включают в себя поражение клубочков и канальцев, аномалии почек и мочевыводящих путей, уролитиаз, множественные кисты, злокачественные или доброкачественные опухоли. Для определения фенотипа нефропатии обычно достаточно результатов общего анализа мочи (гломерулопатия), биохимического анализа крови и мочи, оценки содержания электролитов и кислотно-щелочного равновесия (канальцевые нарушения), ультразвукового исследования и/или компьютерной и магнитно-резонансной томографии (кисты, опухоли, гипоплазия почек, камни). Хроническая почечная недостаточность осложняет течение любого хронического заболевания почек, однако сроки ее развития отличаются. Например, инфантильный нефропатический цистиноз является одной из основных причин диализзависимой хронической почечной недостаточности у детей, в то время как при нефропатии Фабри заместительную почечную терапию обычно приходится начинать в возрасте 30-50 лет.
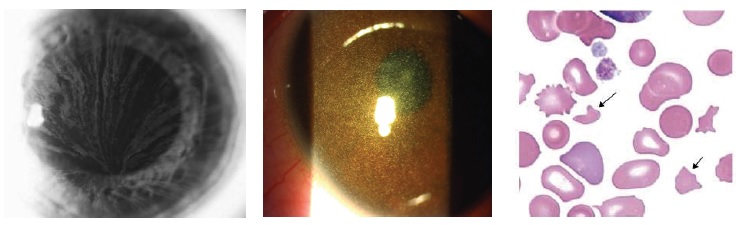
Внепочечные проявления. Поражение почек может быть единственным или ведущим проявлением наследственного заболевания, однако у многих пациентов могут быть выявлены те или иные системные проявления, наличие которых имеет важное диагностическое значение (рис. 1), например, глухота при синдроме Альпорта, нейропатическая боль, ангиокератомы и вихревидная кератопатия при болезни Фабри, светобоязнь и отложение кристаллов цистина в роговице при нефропатическом цистинозе, полиневропатия и поражение сердца при наследственном ATTR-амилоидозе, подагра при аутосомно-доминантной тубулоинтерстициальной болезни почек. При болезнях накопления отложение патологических веществ в тканях происходит постепенно, поэтому частота поражения различных внутренних органов увеличивается с возрастом.
Подтверждение диагноза. При наследственных ор фан ных заболеваниях, характеризующихся поражением клубочков и/или тубулоинтерстиция (микро- или макрогематурия, протеинурия, нефротический синдром и/или нарастание сывороточного уровня креатинина), обычно проводится биопсия почки. Она позволяет исключить более распространенные варианты поражения почек, прежде всего гломерулонефрит, и выявить отложения амилоида, хотя при наследственных нефропатиях, таких как синдром Альпорта или болезнь Фабри, результаты гистологического исследования нефробиоптата на светооптическом уровне могут оказаться мало информативными (фокальный сегментарный гломерулосклероз). В таких случаях для подтверждения диагноза требуется электронная микроскопия ткани почки. При амилоидозе отложения патологического белка могут быть обнаружены не только в почке, но и в слизистой оболочке двенадцатиперстной или прямой кишки или подкожной жировой клетчатке.
Для диагностики некоторых наследственных заболеваний, поражающих почки, могут быть использованы биохимические тесты, например, увеличение содержания цистина в лейкоцитах при нефропатическом цистинозе, снижение активности a-галактозидазы А и увеличение концентрации глоботриаозилсфингозина (Lyso-GL3) в сухих пятнах крови при болезни Фабри, повышение экскреции оксалата с мочой при первичной гипероксалурии. Для окончательного подтверждения диагноза проводится молекулярно-генетическое исследование с целью выявления патогенных вариантов (мутаций) соответствующих генов.
Альтернативой анализу фенотипа в диагностике редких наследственных заболеваний почек может стать полноэкзомное секвенирование. D. Connaughton и соавт. провели полное секвенирование экзома в 114 семьях пациентов с хронической болезнью почек (ХБП) неизвестной этиологии (n=138) [8]. В целом патогенные варианты генов, ассоциирующиеся с развитием ХБП, были выявлены в 37% случаев. При наличии семейного анамнеза они определялись в 36% случаев, внепочечных проявлений – в 69%, а при отсутствии отягощенного семейного анамнеза и системных проявлений – только в 15%. При некоторых наследственных болезнях, имеющих сходные клинические проявления, например, при моногенных аутовоспалительных заболеваниях (семейных периодических лихорадках), сопровождающихся развитием АА-амилоидоза, возможно не полноэкзомное секвенирование, а определение вариантов наборов (панелей) генов. При анализе генетической информации следует учитывать, что количество описанных мутаций при моногенных заболеваниях может исчисляться десятками или сотнями, а клиническое значение части из них остается неопределенным. В связи с этим наличие мутации гена может быть недостаточным для установления диагноза соответствующего моногенного заболевания.
В последние годы стоимость полноэкзомного секвенирования постоянно снижается, поэтому в ближайшем будущем оно будет рассматриваться как альтернатива исследованию определенных генов или панелей генов, особенно если причиной нефропатии могут быть многочисленные заболевания со сходным клиническим фенотипом.
Методы лечения
Заместительная почечная терапия позволяет на многие годы продлить жизнь пациентам с хронической почечной недостаточностью любой этиологии. Чтобы задержать ее развитие и прогрессирование, при редких наследственных нефропатиях применяют стандартные подходы, включая контроль АД и содержания мочевой кислоты, малобелковую диету, ингибиторы ренинангиотензин-альдостероновой системы и натрий-глюкозного транспортера 2 типа и др. При некоторых редких наследственных заболеваниях возможна специфическая патогенетическая терапия. Наиболее ярким примером является экулизумаб – моноклональное антитело к С5 компоненту комплемента, введение которого позволяет восстановить функцию почек у 80% пациентов с атипичным гемолитико-уремическим синдромом (аГУС), ассоциированным с мутациями генов системы комплемента, в том числе уже начавших лечение гемодиализом [9]. Разработаны патогенетические средства и для лечения нефропатического цистиноза (цистеамин), болезни Фабри (агалсидаза альфа, агалсидаза бета, мигаластат), ATTR-амилоидоза (тафамидис, патисиран), аутосомно-доминантной поликистозной болезни почек (толваптан, пролонгированные формы октреотида), первичной гипероксалурии 1 типа (лумасиран). При некоторых заболеваниях, например, лизосомных болезнях накопления или первичной гипероксалурии, специфическая патогенетическая терапия имеет важное значение для профилактики прогрессирования поражения не только почек, но и других органов. Как указано выше, в ближайшем будущем можно ожидать увеличения количества орфанных препаратов, предназначенных для лечения редких наследственных заболеваний.
Варианты поражения почек при орфанных наследственных заболеваниях
Возможные варианты поражения почек при орфанных наследственных заболеваниях включают в себя цилиопатии, врожденные аномалии почек и мочевыводящих путей, гломерулопатии, злокачественные и доброкачественные опухоли, тубулопатии и поражение тубулоинтерстиция. Отдельно выделяют нефропатии при наследственных метаболических заболеваниях, характеризующихся отложением различных веществ в органах и тканях.
Судить о соотношении клинических фенотипов наследственных нефропатией в общей популяции позволяют данные регистра редких заболеваний почек (RKReg) более чем у 7600 взрослых и детей в 21 стране Европейского Союза [10]. Следует учитывать, что в регистр включают пациентов не только с наследственными нефропатиями, но и редкими аутоиммунными заболеваниями (системные васкулиты, системная красная волчанка и др.) и врожденными аномалиями почек и мочевыводящих путей (congenital anomalies of the kidney and urinary tract – CAKUT). Роль генетических дефектов в развитии последних доказана только в части случаев (около 20%) [11]. У взрослых в структуре редких заболеваний почек преобладали гломерулопатии (55%) и цилиопатии, включая аутосомно-доминантную поликистозную болезнь почек (32%), а у детей – врожденные аномалии почек и мочевыводящих путей (34%) и гломерулопатии (31%). Диагноз основного заболевания был подтвержден при молекулярно-генетическом исследовании у 24% детей и 12% взрослых, а диагноз гломерулопатии при биопсии почки – у 39% и 80%, соответственно.
Цилиопатии. Группа заболеваний, в основе которых лежит наследственно обусловленный дефект структуры двигательных ресничек (цилий) клеток почек и других органов. В настоящее время известно более 35 цилиопатий, ассоциирующихся с мутациями около 180 генов, кодирующих синтез цилиарных белков [12]. Наиболее распространенным вариантом цилиопатий, поражающих почки, у взрослых и детей является аутосомно-доминантная и аутосомно-рецессивная поликистозная болезнь почек, соответственно, характеризующаяся образованием множественных кист в почечной паренхиме [13]. Причиной кистозной перестройки ткани почек, которая в конечном итоге приводит к развитию хронической почечной недостаточности, являются повышенная пролиферация и дифференцировка эпителия канальцев и усиление секреции ими жидкости [14]. Данные о распространенности аутосомно-доминантной поликистозной болезни почек в популяции варьируются от >1:500 до 1:2500-4000 [15], поэтому она не является орфанным заболеванием, особенно если ориентироваться на критерии, принятые в Российской Федерации. В то же время аутосомно-рецессивная поликистозная болезнь почек встречается значительно реже – примерно 1:10000 [15].
Причиной аутосомно-доминантной поликистозной болезни почек являются мутации генов PKD1 и PKD2, кодирующих почечные цилиарные белки полицистин 1 и 2, соответственно. Полицистин 1 представляет собой сцепленный с G-белком рецептор, лиганды которого остаются неизвестными, а полицистин 2 – сцепленный с рецептором неселективный ионный канал. Два белка на мембране ресничек образуют комплекс, который регулирует различные клеточные процессы [16].
В основе развития аутосомно-рецессивной полики-стозной болезни почек лежат мутации гена PKHD1,который экспрессируется в основном в почках и вменьшей степени в печени и кодирует трансмембран-ный белок фиброцистин (или полидуктин), регулирую-щий пролиферацию, апоптоз и поляризацию клеток[17].
Для обеих форм поликистозной болезни почек характерно образование множественных кист, которые замещают функционирующую паренхиму и вызывают прогрессирующее увеличение объема почек. Кисты могут быть выявлены при ультразвуковом исследовании и компьютерной или магнитно-резонансной томографии. Клинические симптомы включают в себя арте риальную гипертонию, боль в животе или пояснице, гематурию и инфекции мочевых путей. Аутосомнорецессивная форма заболевания характеризуется более тяжелым течением и развитием диализзависимой хронической почечной недостаточности в детском или подростковом возрасте, в то время как при аутосомнодоминантной форме поликистоз почек обычно выявляют в старшем возрасте. При обеих формах (или заболеваниях) наблюдаются системные проявления, в частности поликистоз печени при аутосомно-доминантной поликистозной болезни почек и дилатация желчных путей, врожденный фиброз печени и портальная гипертензия при аутосомно-рецессивной.
Для лечения аутосомно-доминантной поликистозной болезни почек применяют толваптан, блокирующий V2-рецепторы вазопрессина и снижающий внутриклеточную концентрацию цАМФ. В 13 плацебоконтролируемых клинических исследованиях в целом у 3575 пациентов с аутосомно-доминантной поликистозной болезнью почек толваптан замедлял снижение скорости клубочковой фильтрации (СКФ) и увеличение общего объема почек, снижал риск осложнений, таких как боль, инфекции мочевых путей, гематурия и артериальная гипертония. Основными нежелательными эффектами препарата были жажда, полиурия и повреждение печени [18]. При аутосомно-доминантной поликистозной болезни почек перспективным считают также применение длительно действующих аналогов соматостатина (октреотида), в том числе в комбинации с толваптаном [19], в то время как эффективность ингибиторов mTOR (эверолимуса и сиролимуса) не была доказана [13]. Методы лечения аутосомно-рецессивной поликистозной болезни почек не разработаны.
Учитывая вариабельность фенотипической экспрессии аутосомно-доминантной и аутосомно-рецессивной поликистозной болезни почек и различия подходов к их лечению, а также возможность наличия множественных кист в почках при других ненаследственных и наследственных (аутосомно-доминантная тубулоинтерстициальная болезнь почек, медуллярная губчатая почка, нефронофтиз, болезнь фон Гиппель-Линдау, тубе розный склероз и др.) заболеваниях [20], для под тверждения диагноза целесообразно проводить молекулярно-генетическое исследование.
Врожденные аномалии почек и мочевыводящих путей. Нарушения эмбрионального развития, которые приводят к развитию различных пороков, таких как аплазия, гипоплазия или дисплазия почек, везикоуретральный рефлюкс, мегауретер, клапаны задней уретры и др. Врожденные аномалии почек и мочевыводящей системы в 60-85% случаев диагностируют антенатально (прежде всего в третьем триместре беременности) при ультразвуковом исследовании, в остальных – в детском или подростковом возрасте [21]. При некоторых пороках, например, аплазии почек, какие-либо симптомы могут отсутствовать. Выживаемость пациентов с врожденными аномалиями почек и мочевыводящих путей увеличилась благодаря хирургическому лечению, а также заместительной почечной терапии, которая может потребоваться таким больным в молодом возрасте [22]. Доля пороков почек и мочевыводящих путей в структуре причин диализзависимой хронической почечной недостаточности, развившейся в течение первых 30 лет жизни, составляет примерно 40% [23]. В настоящее время известны сотни генов-кандидатов (PAX2, HNF1β, SIX1, SIX5, SALL1, EYA1, GATA3, FRAS1, FREM2 и др.), мутации которых гипотетически могут быть причиной врожденных аномалий почек и мочевыводящих путей, однако на практике моногенное их происхождение удается подтвердить не более чем в 20% случаев [22]. Пока результаты молекулярно-генетического исследования не влияют на тактику лечения и, соответственно, представляют интерес только с научной точки зрения, хотя в некоторых случаях могут иметь значение для планирования семьи.
Опухоли почки. Злокачественные опухоли почек в 5-8% случаев ассоциированы с редкими наследствен ными заболеваниями, такими как болезнь фон Гип пель-Линдау, туберозный склероз, синдром БертаХогга-Дьюбе, наследственная папиллярная карцинома почки, наследственный лейомиоматоз и почечноклеточный рак, почечноклеточная карцинома с дефицитом сукцинатдегидрогеназы [24]. Болезнь фон Гиппель-Линдау – аутосомно-доминантное наследственное заболевание, обусловленное мутациями гена-супрессора опухолей VHL, которые предрасполагают к развитию злокачественных и доброкачественных опухолей в различных органах, в том числе гемангиобластом головного и спинного мозга и сетчатки, почечноклеточного рака, феохромоцитомы, параганглиомы, нейроэндокринных опухолей поджелудочной железы, опухолей эндолимфатического мешка височной кости, папиллярных цистаденом придатков яичек и широкой связки [25]. Для болезни фон Гиппель-Линдау характерно также наличие множественных кист в обеих почках (обычно они не сопровождаются нарушением их функции) и поджелудочной железе. У 70% пациентов в возрасте до 60 лет развивается почечноклеточный рак, который является одной из главных причин смертности. Лечение хирургическое. Недавно для лечения почечноклеточного рака был зарегистрирован белзутифан – пероральный ингибитор фактора, индуцируемого гипоксией (HIF)-2a [26].
Туберозный склероз – аутосомно-доминантное заболевание, обусловленное мутациями генов TSC1 или TSC2, которые кодируют гамартин и туберин, соответственно. Последние образуют белковый комплекс, регулирующий пролиферацию клеток [27]. Мутации указанных генов сопровождаются стойкой активацией сигнальной системы mTOR, которая приводит к неконтролируемой клеточной пролиферации и развитию множественных доброкачественных опухолей в различных органах. При туберозном склерозе поражаются кожа (гипопиг ментные пятна, ангиофибромы на лице, шагреневые бляшки), головной мозг (субэпендимальная гигантоклеточная астроцитома, корковые туберы), орган зрения (гамартомы сетчатки), сердце (рабдомиомы), легкие (лимфангиолейомиоматоз). Часто встречаются нейропсихические расстройства, такие как судороги, аутизм и нарушение когнитивной функции. Поражение почек характеризуется наличием ангиомиолипом, обнаруживаемых в детском или подростковом возрасте, и кист, в то время как почечноклеточный рак развивается редко. Почечные ангиомиолипомы нередко осложняются кровотечениями, которые могут привести к нарушению функции почек и смерти. Препараты первой линии в лечении туберозного склероза – ингибиторы mTOR, в том числе эверолимус и сиролимус.
Гломерулопатии. Классическим примером наследственной гломерулопатии является синдром Альпорта, который характеризуется сочетанием поражения почек с нейросенсорной тугоухостью и изменениями со стороны органа зрения, включая помутнение роговицы, передний лентиконус (ограниченное выпячивание в передней части хрусталика), точечно-пятнистую ретинопатию, истончение сетчатки [28,29]. Причина заболевания – мутации генов COL4A3, COL4A4 и COL4A5, кодирующих α3, α4 и α5-цепи коллагена IV типа, который входит в состав базальных мембран клубочков, улитки внутреннего уха, хрусталика, сетчатки и роговицы глаза. Основной путь наследования – сцепленный с Х-хромосомой, реже заболевание передается по аутосомно-доминантному или аутосомно-рецессивному типу. У мужчин с Х-сцепленным вариантом синдрома Альпорта заболевание протекает тяжелее, чем у женщин, у которых мутантный ген находится в гетерозиготном положении. Первый признак поражения почек при синдроме Альпорта – микрогематурия, которая развивается в детском возрасте, позднее появляются альбуминурия/протеинурия и нарушение функции почек. У мужчин с Х-сцепленным синдромом Альпорта заместительную почечную терапию обычно приходится начинать в возрасте 30-40 лет. Изменения при светооптической микроскопии почечного биоптата при синдроме Альпорта отсутствуют или неспецифичны (фокальный сегментарный гломерулосклероз), поэтому важное диагностическое значение имеют результаты электронной микроскопии, которая позволяет выявить истончение базальной мембраны клубочков, а позднее ее очаговое, а затем диффузное утолщение и расслоение и подоцитопатию. При иммуногистохимическом исследовании можно обнаружить снижение экспрессии α5-цепи коллагена IV в биоптате почки и кожи. Для профилактики прогрессирующего ухудшения функции почки пациентам с синдромом Альпорта показана ранняя нефропротективная терапия ингибиторами АПФ или блокаторами ангиотензиновых рецепторов, а также ингибиторами натрийглюкозного котранспортера 2 типа.
Сегодня известно еще несколько десятков генов (NPHS1, NPHS2, INF2, TRPC6, ACTN4, CD2AP, MYO1E и др.), мутации которых приводят к развитию наследственных гломерулопатий, обычно проявляющихся стероидорезистентным нефротическим синдромом в детском, подростковом или молодом возрасте и характеризующихся картиной фокального сегментарного гломерулосклероза (ФСГС) или реже минимальными изменениями или диффузным мезангиальным склерозом при биопсии почки [30,31]. У части больных с генетическим ФСГС нефротический синдром отсутствует, но наблюдается протеинурия, сочетающаяся или не сочетающаяся с нарушением функции почек. Поражение почек может быть изолированным или сопровождается различными внепочечными проявлениями. Мутации генов, передающиеся по аутосомнорецессивному типу, обычно вызывают развитие стероидорезистентного нефротического синдрома в детском возрасте, а варианты генов, которые наследуются по аутосомно-доминантному типу, – у взрослых.
Причиной моногенных гломерулопатий являются мутации генов, кодирующих структурные или сигнальные белки, которые обеспечивают целостность и функцию фильтрационного барьера клубочков почек, состоящего из подоцитов, гломерулярной базальной мембраны и эндотелия. Мутации генов могут затрагивать белки щелевой диафрагмы (нефрин, подоцин, CD2-ассоциированный белок и др.), структурные и регуляторные белки цитоскелета (a-актинин 4, инвертированный формин 2, аниллин и др.), белки адгезии (интегрины, ламинин), белки базальной мембраны клубочков (коллаген IV типа, ламинин α5), ядерные факторы транскрипции (WT1, SMARCA-подобный белок), белки ядерных поровых комплексов (нуклеопорины, экспортин 5), белки, участвующие в биосинтезе коэнзима Q10, и др. (кубилин, фосфоманномутаза 2, диацилглицеролкиназа) [32]. У взрослых, наряду с мутациями гена COLА3/А4/А5, частой причиной наследственной гломерулопатии являются мутации гена NPHS2, кодирующего подоцин, реже встречаются мутации генов INF2 (инвертированный формин 2), TRPC6 (transient receptor potential canonical 6), ACTN4 (a-актинин 4) и др. Как указано выше, мутации некоторых генов вызывают развитие гломерулопатии, сочетающейся с различными внепочечными проявлениями. Помимо синдрома Альпорта примерами могут служить синдром ногтей-надколенника (мутации LMX1B; дисплазия ногтей и надколенника, глаукома), синдром Шимке (мутации SMARCAL1; низкий рост, спондило эпифизарная дисплазия, нарушение клеточного иммунитета, артериопатия), WT1-ассоциированные синдромы (мутации WT1), митохондриопатии (мутации COQ2, PDSS2, COQ6 и COQ8; неврологические и поведенческие расстройства и др.), синдром Пирсона (мутации LAMB2; микрокория, изменения сетчатки и др.) [30].
Генетическую природу гломерулопатии удается установить примерно у 30% детей и 10-15% взрослых со стероидорезистентным нефротическим синдромом [33,34]. В прошлом с этой целью тестировали панели генов, однако сегодня чаще проводят полноэкзомное секвенирование. Диагностика моногенных форм ФСГС имеет важное практическое значение, так как позволяет избежать последствий неэффективной иммуносупрессивной терапии и предсказать низкий риск рецидива после трансплантации почки [32]. При митохондропатиях раннее применение коэнзима Q10 может задержать прогрессирующее ухудшение функции почек [35].
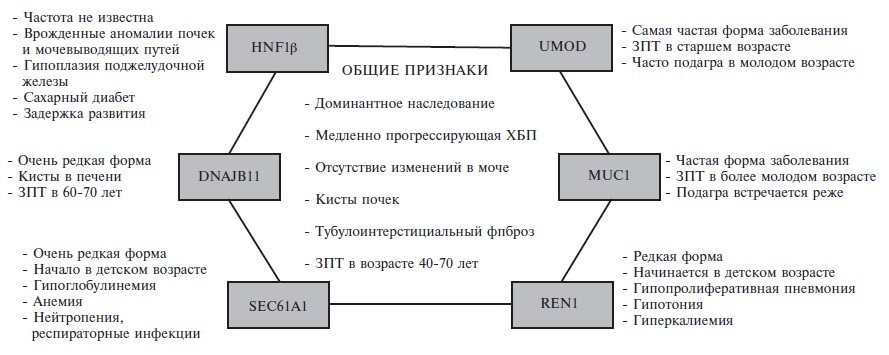
Тубулоинтерстициальные заболевания почек. Одним из наиболее распространенных вариантов моногенных заболеваний почек (после аутосомно-доминантной поликистозной болезни почек и синдрома Альпорта) считают аутосомно-доминантную тубулоинтерстициальную болезнь почек, которая может быть обусловлена мутациями различных генов, в том числе UMOD (уромодулин), MUC1 (муцин 1), REN (препроненин), HNF1B (гепатоцитарный ядерный фактор 1β), SEC61A1 (&alha;1-субъединица SEC61 транслокона) и DNAJB11 (кофактор GRP78/BiP). Общими признаками для всех типов аутосомно-доминантной тубулоинтерстициальной болезни почек являются медленное нарастание сывороточного уровня креатинина, развитие диализзависимой хронической почечной недостаточности в возрасте от 40 до 70 лет, отсутствие гематурии и других изменений в моче (может наблюдаться небольшая протеинурия), кисты в почках, не сопровождающиеся значительным увеличением их размеров, и тубулоинтерстициальный фиброз при гистологическом исследовании биоптата почки [36,37]. Особенности течения заболевания, в том числе возраст, в котором начинается ухудшение функции почек, и наличие внепочечных проявлений, зависят от типа мутации (табл. 1) [36]. Важную роль в патогенезе аутосомно-доминантной тубулоинтерстициальной болезни почек играет образование несвернутых или неправильно свернутых протеинов, которые вызывают стресс эндоплазматического ретикулума и цитотоксичность. Наиболее распространенная форма аутосомно-доминантной тубулоинтерстициальной болезни почек обусловлена мутациями гена UMOD, кодирующего уромодулин. У 70-80% таких пациентов имеется подагра, которая развивается в молодом возрасте, в том числе у женщин, и предшествует появлению признаков ХБП. Гиперурикемия и подагра наблюдаются также (хотя и реже) и при варианте заболевания, обусловленном мутациями гена MUC1, который кодирует муцин. Другие формы аутосомнодоминантной тубулоинтерстициальной болезни почек встречаются редко или очень редко. Специфических методов лечения не существует. При наличии гиперурикемии важное значение для профилактики прогрессирования ХБП имеет применение гипоурикемических препаратов (аллопуринола или фебуксостата).
| Заболевание | Препарат | Механизм действия |
|---|---|---|
| Аутосомно-доминантная | Толваптан | Блокатор V2-рецепторов вазопрессина |
| поликистозная болезнь почек | Октреотид | Аналог соматостатина |
| Болезнь фон Гиппель-Линдау | Белзутифан | Ингибитор фактора, индуцируемого гипоксией (HIF-2α) |
| Туберозный склероз | Эверолимус, сиролимус |
Ингибиторы mTOR |
| Нефропатический цистиноз | Цистеамин | Обеспечивает выведение цистина из лизосом независимо от цистинозина |
| Болезнь Фабри | Агалсидаза альфа,агалсидаза бета | Рекомбинантные препараты a-галактозидазы А |
| Мигаластат | Шаперон, стабилизирующий активность α-галактозидазы А | |
| ATTR-амилоидоз | Тафамидис | Стабилизирует транстиретин и препятствует образованию амилоидогенных мономеров |
| Патисаран | Олигонуклеотид, подавляющий синтез транстиретина путем РНК-интерференции | |
| Первичная оксалурия | Лумасиран | Ингибирование мРНК гена гидроксикислой оксидазы 1, кодирующего гликолатоксидазу, и уменьшение содержания доступного глиоксилата в печени. |
| аГУС | Экулизумаб | Моноклональное антитело, взаимодействующее с С5 компонентом комплемента и блокирующее образование анафилотоксина С5а и мембраноатакующего комплекса |
Тубулопатии. Наследственные тубулопатии включают в себя около 50 генетических заболеваний, характеризующихся нарушением функции канальцевого аппарата почек на различных уровнях [38]. Соответственно, выделяют проксимальные, петлевые и дистальные тубулопатии. Клинические проявления тубулопатий неспецифичные. Могут наблюдаться полиурия, полидипсия, раздражительность, нарушение роста у детей и изменения АД, а также небольшая протеинурия (за счет низкомолекулярных белков). Важное диагностическое значение имеют определение кислотно-щелочного равновесия, электролитов в сыворотке крови и моче, активности ренина и альдостерона при наличии гипоили гиперкалиемии, экскреции β2-микроглобулина с мочой (маркер канальцевой дисфункции), а также анализ возможных внепочечных проявлений и изучение семейного анамнеза [39]. Наследственные тубулопатии обычно проявляются в детском или подростковом возрасте, в то время как у взрослых повреждение канальцев чаще является приобретенным (лекарственные средства, интоксикации тяжелыми металлами, злокачественные новообразования, в том числе гематологические неоплазии, и др.).
Гипокалиемический метаболический ацидоз может быть следствием потери бикарбонатов в проксимальных канальцах (проксимальный почечный канальцевый ацидоз [ПКА], 2 тип) или нарушения секреции ионов водорода в дистальных канальцах (дистальный ПКА, 1 тип). Генерализованная дисфункция проксимальных почечных канальцев (почечный синдром Фанкони) сопровождается увеличением экскреции с мочой не только бикарбонатов (ПКА 2 типа), но и аминокислот, глюкозы, фосфора, натрия, калия, кальция, мочевой кислоты и карнитина, которые в норме реабсорбируются в этом отделе канальцевого аппарата [40,41]. Помимо задержки роста, характерное проявление синдрома Фанкони в детском возрасте – гипофосфатемический рахит. У детей основной причиной синдрома Фанкони является нефропатический цистиноз (см. ниже), более редкими – болезнь Вильсона-Коновалова, наследственная тирозинемия, наследственная непереносимость фруктозы, галактоземия 1 типа и синдром ФанкониБикеля, при которых проксимальную канальцевую дисфункцию связывают с внутриклеточным накоплением меди, тирозина, фруктозы, галактозы и гликогена, соответственно [42]. Неполная дисфункция проксимальных канальцев, сочетающаяся с разнообразными внепочечными проявлениями, наблюдается при болезни Гента 1 (мутации CLCN5) и 2 типа (мутации OCRL), синдроме Лоу (мутации OCRL) и изолированном проксимальном ПКА 2 типа (мутации SLC4A4). Нарушение секреции ионов водорода в дистальных канальцах, сопровождающееся потерей калия с мочой, отмечается при дистальном ПКА 1 типа (мутации ATP6V1B1, ATP6V0A4, FOXI1, SLC4A1 и WDR72).
Гипокалиемический метаболический алкалоз. У детей с нормальным АД или артериальной гипотонией основными причинами гипокалиемического гипохлоремического алкалоза являются синдромы Барттера и Гительмана, связанные с нарушением транспорта натрия в петле Генле и дистальных канальцах, соответственно. Отличительными признаками синдрома Гительмана являются развитие в детском или подростковом возрасте, клинически явная гипомагнезиемая, сопровождающаяся судорогами в мышцах, и гипокальциурия, в то время как для синдрома Барттера, развивающегося у новорожденных или маленьких детей, характерны массивная полиурия и гиповолемия, гиперкальциурия и нефрокальциноз [43].
Причинами гипокалиемического метаболического алкалоза, сочетающегося с артериальной гипертонией, могут быть семейный гиперальдостеронизм 1-4 типа и синдром Лиддля (псевдоальдостеронизм) [44]. Последний развивается в результате мутаций генов SCNN1A/B, которые кодируют a и β-субъединицы амилоридчувствительных эпителиальных натриевых каналов (ENaC) в собирательных трубках. Активация этих каналов приводит к реабсорбции воды и натрия и потере калия независимо от альдостерона, поэтому активность гормона в плазме при синдроме Лиддля подавлена, в отличие от первичного гиперальдостеронизма.
Гиперкалиемический метаболический ацидоз. Раз ви вается при псевдогиперальдостеронизме, который может быть следствием резистентности к минералокортикоидам или активации/нарушения деградации тиазид чувствительного натрий-хлор котранспортера в почечных канальцах.
Гипернатриемия/гипонатриемия. Могут быть следствием нефрогенного несахарного диабета (уменьшение объема циркулирующей жидкости, гипернатриемия) или синдрома неадекватной продукции антидиуретического гормона (увеличение объема циркулирующей жидкости, гипонатриемия разведения).
Почечная глюкозурия. Редкая тубулопатия, приводящая к повышенной экскреции глюкозы с мочой при нормальной концентрации глюкозы в крови. Как правило протекает бессимптомно и не требует лечения (доброкачественная глюкозурия) [45]. Иногда сопровождается полиурией и рецидивирующими инфекциями мочевыводящих путей. Почечная глюкозурия связана с мутацией гена SLC5A2, кодирующего натрийглюкозный котранспортер 2 типа (SGLT2) в проксимальных канальцах. SGLT2 является основным котранспортером глюкозы в проксимальных канальцах, ответственным за 90% реабсорбции глюкозы в почках.
Метаболические нарушения. Поражение почек часто встречается при наследственных заболеваниях, сопровождающихся отложением в тканях различных веществ, например, гликосфинголипидов при болезни Фабри, цистина при нефропатическом цистинозе, оксалатов при первичной гипероксалурии.
Нефропатический цистиноз обусловлен накоплением кристаллов цистина в лизосомах клеток почек и других органов в результате мутаций гена CTNS и дефицита транспортного белка цистинозина [46]. Заболевание проявляется генерализованной дисфункцией проксимальных почечных канальцев (синдром Фанкони), которая развивается на первом году жизни [47]. Позднее начинается снижение скорости клубочковой фильтрации (СКФ), которое приводит к формированию терминальной стадии ХБП в возрасте 8-12 лет. При более редкой ювенильной форме заболевания функция почек ухудшается медленнее, а диализзависимая хроническая почечная недостаточность развивается в старшем возрасте (от 12 до 28 лет) [48].
Первое внепочечное проявление цистиноза – отложение кристаллов цистина в роговице, которое определяется к возрасту 16 мес у всех пациентов и имеет важное диагностическое значение. Первоначально отложение цистина не сопровождается симптомами, однако в возрасте 8-12 лет появляется светобоязнь. В старшем возрасте у пациентов развиваются другие системные проявления, в том числе гипотиреоз, сахарный диабет, гипогонадизм (у мужчин), миопатия с поражением мышц конечностей и дыхательных мышц, неврологические расстройства.
Предполагать нефропатический цистиноз следует у всех больных, получающих заместительную почечную терапию с детского или подросткового возраста, особенно при наличии анамнестических указаний на синдром Фанкони в первые годы жизни и светобоязнь. Скрининговым методом является осмотр роговицы с помощью щелевой лампы, который позволяет выявить отложения цистина. Для подтверждения диагноза измеряют концентрацию цистина в лейкоцитах и проводят молекулярно-генетическое исследование.
Для лечения нефропатического цистиноза применяют цистеамина битартрат, который проникает в лизосомы, расщепляет цистин на две молекулы цистеина и соединяется с одной из них с помощью дисульфидного мостика. Цистеин и комплекс цистеина и циастеамина не нуждаются в цистозине для выхода из лизосом, а лечение цистеамином вызывает выведение цистина из лизосом клеток и предупреждает дальнейшее его накопление. Препарат выпускается в виде капсул для приема внутрь и глазных капель, содержащих 0,55% раствор цистеамина гидрохлорида. Капли используют для растворения кристаллов цистина в роговице, так как пероральная терапия цистеамином не оказывает на них влияние. Лечение цистеамином позволяет задержать развитие терминальной хронической почечной недостаточности на 6-10 лет и предупреждает или тормозит развитие внепочечных проявлений цистиноза [49]. В когортном исследовании у больных нефропатическим цистинозом, продолжавших терапию цистеамином более 20 лет, частота сахарного диабета и миопатии снизилась с 28% до 0% и с 60% до 0%, соответственно, а у больных, получавших препарат в течение более 8 лет, частота гипотиреоза снизилась с 87% до 56% [50].
Болезнь Фабри – лизосомная болезнь накопления, связанная с мутациями гена GLA, расположенного на Х-хромосоме и характеризующаяся накоплением гликосфинголипидов в различных органах и тканях в результате снижения или полного отсутствия активности лизосомного фермента a-галактозидазы А. При классическом варианте БФ первые симптомы, в том числе нейропатическая боль (акропарестезии), ангиокератомы и сниженное потоотделение, появляются в детском или подростковом возрасте, а поражение почек, сердца и головного мозга развивается в возрасте 20-40 лет и более. У части пациентов “классические" симптомы БФ отсутствуют, а первыми проявлениями заболевания в старшем возрасте оказываются гипертрофия левого желудочка неясного происхождения, нефропатия и/или инсульт, развившийся в сравнительно молодом возрасте. Признаки поражения почек (небольшая протеинурия, обычно не достигающая нефротического уровня и постепенное снижение СКФ) отсутствуют у детей и подростков, но наблюдаются у большинства взрослых пациентов с болезнью Фабри и могут потребовать заместительной почечной терапии, прежде всего у мужчин, у которых заболевание характеризуется более тяжелым течением, чем у женщин, за счет Х-сцепленного типа наследования [51].
Предполагать болезнь Фабри у пациентов с нефропатией неясного генеза следует при наличии семейного анамнеза, системности поражения внутренних органов и типичных симптомов, таких как ангиокератомы, нейропатическая боль и вихревидная кератопатия. Результаты гистологического исследования биоптата почки часто трактуются как фокальный сегментарный гломерулосклероз, а для подтверждения диагноза требуется электронная микроскопия образцов ткани, позволяющая выявить типичные “зебровидные" включения. С целью диагностики болезни Фабри измеряют активность a-галактозидазы А (у мужчин) и Lyso-GL3 в сухих каплях крови и проводят молекулярно-генетическое исследование. Эффектив ным методом ранней диагностики заболевания является обследование родственников пробанда (семейный скрининг), которое обычно позволяет выявить еще несколько случаев болезни Фабри в семье, в том числе у детей. Кроме того, возможен скрининг в “группах риска", в которых вероятность выявления болезни Фабри выше, чем в общей популяции. К ним относят пациентов, получающих заместительную почечную терапию, пациентов с гипертрофической кардиомиопатией и инсультом, развившимся в молодом возрасте.
Предполагать болезнь Фабри у пациентов с нефропатией неясного генеза следует при наличии семейного анамнеза, системности поражения внутренних органов и типичных симптомов, таких как ангиокератомы, нейропатическая боль и вихревидная кератопатия. Результаты гистологического исследования биоптата почки часто трактуются как фокальный сегментарный гломерулосклероз, а для подтверждения диагноза требуется электронная микроскопия образцов ткани, позволяющая выявить типичные “зебровидные" включения. С целью диагностики болезни Фабри измеряют активность a-галактозидазы А (у мужчин) и Lyso-GL3 в сухих каплях крови и проводят молекулярно-генетическое исследование. Эффектив ным методом ранней диагностики заболевания является обследование родственников пробанда (семейный скрининг), которое обычно позволяет выявить еще несколько случаев болезни Фабри в семье, в том числе у детей. Кроме того, возможен скрининг в “группах риска", в которых вероятность выявления болезни Фабри выше, чем в общей популяции. К ним относят пациентов, получающих заместительную почечную терапию, пациентов с гипертрофической кардиомиопатией и инсультом, развившимся в молодом возрасте.
Основной метод лечения болезни Фабри – ферментозаместительная терапия рекомбинантными препаратами α-галактозидазы А – агалсидазой альфа (0,2 мг/кг) и агалсидазой бета (1 мг/кг) [52]. В контролируемых и неконтролируемых исследованиях лечение этими препаратами, особенно начатое на более ранней стадии (т.е. при отсутствии необратимых изменений внутренних органов), вызывало уменьшение нейропатической боли, задерживало ухудшение функции почек и увеличение массы миокарда левого желудочка, улучшало показатели качества жизни и исходы заболевания [53].
Амилоидоз. Наследственным вариантом амилоидоза является ATTR-амилоидоз, обусловленный мутациями гена TTR, кодирующего синтез транстиретина. Последний выполняет функции транспортного белка тироксина и витамина А. В результате мутаций синтезируется белок, который не способен образовывать тетрамеры и обладает очень высокой амилоидогенностью [54]. Основные проявления ATTR-амилоидоза – поражение сердца (по типу рестриктивной кардиомиопатии) и/или периферической нервной системы (прогрессирующая симметричная дистальная полиневропатия), однако у части больных наблюдается и поражение почек. Для лечения ATTR-амилоидоза применяют тафамидис, который связывается с транстиретином, стабилизирует его четвертичную структуру и препятствует образованию амилоидогенных мономеров, что позволяет замедлить прогрессирование заболевания [55].
В отличие от ATTR-амилоидоза, поражение почек (нарастающая протеинурия при отсутствии изменений мочевого осадка, нефротический синдром, диализзависимая хроническая почечная недостаточность) является ведущим в клинической картине АА-амилоидоза. Предшественником АА-амилоида является сывороточный амилоидный А-протеин (SAA) – белок острой фазы, содержание которого в крови повышается при любых воспалительных процессах. Хотя АА-амилоидоз не относится к наследственным заболеваниям, он может осложнить течение редких моногенных аутовоспалительных заболеваний, которые в зарубежной литературе называют “семейными периодическими лихорадками". Они включают в себя периодическую болезнь (семейную средиземноморскую лихорадку), криопирин-ассоциированный периодический синдром (CAPS), периодический синдром, ассоциированный с мутацией рецептора фактора некроза опухоли (TRAPS), и синдром недостаточности мевалонаткиназы/гипериммуноглобулинемию D (HIDS/MKD) [56]. Предпо лагать аутовоспалительное заболевание следует при наличии клинических (прежде всего лихорадки) и лабораторных (повышение СОЭ и содержания С-реактивного белка, нейтрофильный лейкоцитоз) признаков воспаления, которые нельзя объяснить другими более частыми причинами, такими как инфекции, злокачественные новообразования или аутоиммунные заболевания. Для семейных периодических лихорадок характерны эпизоды (приступы) лихорадки, сопровождающейся разнообразными кожными высыпаниями, болями в суставах и мышцах, серозитом (перитонит, плеврит, перикардит и артрит) [57]. Приступы часто начинаются в детском или подростковом возрасте, а похожие проявления могут наблюдаться у родственников пациента. При опросе взрослых пациентов обращают на себя внимание длительный анамнез (месяцы и годы, а иногда даже десятилетия), стереотипность приступов и отсутствие симптомов в межприступные периоды. Иногда именно АА-амилоидоз с поражением почек оказывается первым проявлением, заставляющим обсуждать диагноз аутовоспалительного заболевания [58]. При появлении протеинурии у пациента с семейной периодической лихорадкой необходимо во всех случаях исключать диагноз АА-амилоидоза путем гистологического исследования почечного биоптата (окраска конго-рот и поляризационная микроскопия). У части пациентов отложения амилоида можно выявить в биоптате слизистой оболочки двенадцатиперстной или прямой кишки или подкожной жировой клетчатки.
Специфических методов лечения АА-амилоидоза не существует, однако подавление воспаления позволяет предупредить его развитие и затормозить прогрессирование поражения почек. У пациентов с периодической болезнью в большинстве случаев эффективен колхицин, вызывающий значительное снижение частоты или полное прекращение приступов заболевания. Одним из основных медиаторов воспалительного ответа при аутовоспалительных заболеваниях является интерлейкин-1, ингибиторы которого (канакинумаб, анакинра) оказались эффективными при многих заболеваниях этой группы. В двойном слепом, плацебо-контролируемом исследовании CLUSTER, в которое были включены 181 пациент с семейными периодическими лихорадками, частота полного ответа на лечение канакинумабом составила 71% у пациентов с колхицинрезистентной периодической болезнью, 57% – с недостаточностью мевалонаткиназы и 73% – с TRAPS [59]. При этом лечение канакинумабом вызывало значительное снижение содержания SAA, что имеет важное значение для профилактики АА-амилоидоза. По данным мета-анализа 5 исследований, при лечении канакинумабом полный ответ был достигнут у 85,2% из 128 пациентов с криопирин-ассоциированным периодическим синдромом, причем препарат был эффективным как при легком (семейной холодовой крапивнице), так и более тяжелых (синдроме Макла-Уэллса и NOMID) фенотипах заболевания [60].
Первичная оксалурия. Группа аутосомно-рецессивных заболеваний, характеризующихся увеличением продукции оксалата, который образуется в печени из глиоксилата и выводится почками. В высоких концентрациях оксалат образует кристаллы с кальцием в почечных канальцах, что приводит к развитию тубулоинтерстициального воспаления, нефролитиаза и/или нефрокальциноза и в конечном итоге прогрессирующей хронической почечной недостаточности. При снижении СКФ менее 30-40 мл/мин/1,73 м2 оксалат начинает накапливаться в различных тканях (оксалоз), включая кости, сердце, сосуды, нервы и глаза, так как его продукция в печени превышает возможность почечной экскреции [61]. Выделяют три варианта первичной гипероксалурия, которые обусловлены мутациями разных генов (AGXT, GRHPR и HOGA1), кодирующих ферменты метаболизма глиоксилата. Первичная гипероксалурия 1 типа обычно проявляется в детском возрасте и характеризуется более тяжелым течением, чем первичная гипероксалурия 2 и 3 типов, в частности чаще и быстрее приводит к развитию диализзависимой хронической почечной недостаточности, хотя заболевание может быть диагностировано в любом возрасте. Предполагать первичную гипероксалурию как у детей, так и у взрослых следует при наличии рецидивирующего нефролитиаза и/или нефрокальциноза, особенно сопровождающихся прогрессирующим ухудшением функции почек. Скрининговым тестом у пациентов с нормальной функцией почек или ХБП 1-3 стадии является двукратное определение экскреции оксалата с мочой (в разовой порции в пересчете на креатинин или за сутки), в то время как у пациентов с ХБП 4-5 стадии целесообразно измерение содержания оксалата в плазме [62]. При наличии гипероксалурии, которую нельзя объяснить какими-либо другими причинами (употребление продуктов с большим содержанием оксалата и др.), показано молекулярно-генетическое исследование.
Консервативная терапия первичной гипероксалурии предполагает гидратацию (3,5-4 л/сут у взрослых и 2–3 л/м2 у детей) и прием калия цитрата (0,1-0,15 г/кг) [62]. У части пациентов с гипероксалурией 1 типа эффективен пиридоксин (витамин В6). Критерием ответа на лечение этим витамином в течение по крайней мере 3 мес является снижение экскреции оксалата с мочой более чем на 30%. Препарат следует назначать в дозе не более 5 мг/кг, так как эффективность более высоких доз не доказана [62]. У пациентов с ХБП 5 стадии, не отвечающих на пиридоксин, возможна одновременная или последовательная трансплантация печени, являющейся источником оксалата, и почек, что позволяет избежать прогрессирования оксалоза в посттранс плантационном периоде.
Для лечения первичной гипероксалурии 1 типа недавно одобрен лумасиран, ингибирующий матричную РНК гена гидроксикислой оксидазы 1, кодирующего гликолатоксидазу, и уменьшающий содержание доступного глиоксилата в печени. В клинических исследованиях 3 фазы применение этого препарата вызывало значительное снижение экскреции оксалата с мочой или его содержания в плазме у детей и взрослых пациентов с нормальной или сниженной функцией почек, соответственно [63,64].
Тромботическая микроангиопатия (ТМА). ТМА – это клинико-морфологический синдром, характеризую щийся повреждением эндотелия и образованием тром бов в сосудах микроциркуляторного русла с их окклюзией и ишемией органов, прежде всего почек [64]. Клинические проявления ТМА – острое повреж дение почек (ОПП), тромбоцитопения и микроангио патическая гемолитическая анемия, развивающаяся в результате механического гемолиза эритроцитов (Кумбс-негативная) и сопровождающаяся повышением активности лактатдегидрогеназы (ЛДГ) и снижением уровня гаптоглобина в сыворотке крови. Микрососуды почек наиболее уязвимы к активации системы компле мента за счет особого строения (фенестрация) эндоте лия гломерулярных капилляров, поэтому ОПП относится к основным проявлениям аГУС, хотя при этом заболевании могут поражаться и другие органы, в том числе сердце, головной мозг, легкие.
Одной из причин первичной ТМА является аГУС – редкое заболевание, сопровождающееся неконтроли руемой активацией комплемента вследствие мутаций генов регуляторных белков или компонентов компле мента или образования антител к фактору Н системы комплемента. Триггерами развития аГУС часто служат различные комплементактивирующие состояния, в частности в акушерской практике ими являются ослож нения течения беременности (преэклампсия, крово течение и оперативные вмешательства). Диагноз аГУС следует предполагать в случае развития ОПП неясной природы в сочетании с типичными лабораторными изменениями. У 40-60% пациентов с аГУС не удается выявить патогенные мутации генов, регулирующих функцию системы комплемента, поэтому молекулярно генетическое исследование не считают необходимым для подтверждения диагноза, хотя оно имеет важное значение для оценки риска рецидивов заболевания, в том числе после трансплантации почки, и, соответ ственно, для определения необходимости в поддержи вающей комплементблокирующей терапии. Пункцион ная биопсия почки, как и любое другое инвазивное исследование, может вызвать дополнительную актива цию системы комплемента. Тем не менее, она обосно вана, если диагноз вызывает сомнение, в частности при отсутствии полного симптомокомплекса ТМА [66].
Для лечения аГУС помимо плазмообмена применяют экулизумаб – моноклональное антитело, взаимо действующее с С5 компонентом комплемента и бло кирующее образование анафилотоксина С5а и мембраноатакующего комплекса, что позволяет пред отвратить дальнейшее повреждение эндотелия сосудов и подавить образование микротромбов. В клинических исследованиях было показано, что лечение экулизума бом значительно снижает потребность в гемодиализе или трансплантации почки у пациентов с аГУС, в том числе уже начавших заместительную почечную тера пию, а длительное применение этого препарата позво ляет избежать рецидивов заболевания [9].
Заключение
Своевременная диагностика редких наследственных заболеваний почек имеет важное значение для генети ческой консультации и обследования родственников пробанда с целью выявления других случаях болезни в семье. Кроме того, постепенно увеличивается количе ство лекарственных препаратов, которые могут быть использованы для патогенетической терапии орфанных заболеваний (табл. 1). Предполагать наследственный генез нефропатии следует в первую очередь в случае ее развития в детском, подростковом или молодом возрас те, хотя некоторые наследственные заболевания про являются в пожилом возрасте, а семейный анамнез может отсутствовать, в частности при аутосомно-рецес сивном типе наследования. Дифференцировать многие наследственные болезни почек со сходным фенотипом на основании особенностей клинической картины и лабораторных проявлений часто сложно или невозмож но, поэтому диагноз следует подтвердить с помощью молекулярно-генетического метода. Иногда фенотип болезни, например, внепочечные проявления, или результаты скрининговых тестов указывают на конкрет ный диагноз, что позволяет ограничиться тестировани ем определенных генов или панелей генов, хотя в последние годы при подозрении на наследственные заболевания все чаще проводят полноэкзомное секве нирование, стоимость которого существенно снизилась.
Используемые источники
- Joly D, Béroud C, Grünfeld JP. Rare inherited disorders with renal involvementapproach to the patient. Kidney Int 2015;87(5):901-8.
- Nguengang Wakap S, Lambert DM, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet 2020;28(2):165-73.
- Рамеев В.В., Симонян А.Х., Богданова М.В. и др. Периодическая болезнь: эволюция представлений о заболевании и подходы к диагностике и лечению. Клин фармакол тер 2020;29(2):56-68 [Rameev VV, Simonyan AKh, Bogdanova MV, et al. Familial Mediarranean fever: diagnosis and treatment. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2020;29(2):56-67 (In Russ.)].
- Devuyst O, Knoers NV, Remuzzi G, Schaefer F. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet 2014;383(9931):1844-59.
- Boycott KM, Lau LP, Cutillo CM, Austin CP. International collaborative actions and transparency to understand, diagnose, and develop therapies for rare diseases. EMBO Mol Med 2019;11(5):e10486.
- Granhøj J, Tougaard B, Lildballe DL, Rasmussen M. Family history is important to identify patients with monogenic causes of adult-onset chronic kidney disease. Nephron 2022;146(1):49-57.
- Moiseev S, Tao E, Moiseev A, et al. The benefits of family screening in rare diseases: genetic testing reveals 165 new cases of Fabry disease among at-risk family members of 83 index patients. Genes (Basel) 2022;13(9):1619.
- Connaughton DM, Kennedy C, Shril S, et al. Monogenic causes of chronic kidney disease in adults. Kidney Int 2019;95(4):914-28.
- Козловская Н.Л., Моисеев С.В. Комплементблокирующая терапия у пациентов с атипичным гемолитико-уремическим синдромом и вторичной тромботической микроангиопатией. Клин фармакол тер 2023;22(2):7-14 [Korotchaeva Yu, Kozlovskaya N, Moiseev S. Complement inhibition therapy in patients with atypical hemolytic uremic syndrome and secondary microangiopathies. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2023;32(2):7-14 (In Russ.)].
- Bassanese G, Wlodkowski T, Servais A, et al. The European Rare Kidney Disease Registry (ERKReg): objectives, design and initial results. Orphanet J Rare Dis 2021;16(1):251.
- Chevalier RL. CAKUT: A pediatric and evolutionary perspective on the leading cause of CKD in childhood. Pediatr Rep 2023;15(1):143-53.
- McConnachie DJ, Stow JL, Mallett AJ. Ciliopathies and the kidney: A review. Am J Kidney Dis 2021;77(3):410-9.
- Руденко Т.Е., Бобкова И.Н., Ставровская Е.В. Современные подходы к консервативной терапии поликистозной болезни почек. Терапевтический архив 2019;91(6):116–23 [Rudenko TE, Bobkova IN, Stavrovskaya EV. Modern approaches to conservative therapy of polycystic kidney disease. Therapeutic Archive 2019;91(6):116–23 (In Russ.)].
- Qiu J, Germino GG, Menezes LF. Mechanisms of cyst development in polycystic kidney disease. Adv Kidney Dis Health 2023;30(3):209-219.
- Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers 2018;4(1):50.
- Ma M, Gallagher AR, Somlo S. Ciliary mechanisms of cyst formation in polycystic kidney disease. Cold Spring Harb Perspect Biol 2017;9(11):a028209.
- Sudarikova AV, Vasileva VY, Sultanova RF, Ilatovskaya DV. Recent advances in understanding ion transport mechanisms in polycystic kidney disease. Clin Sci (Lond) 2021;135(21):2521-40.
- Lu J, Xu W, Gong L, et al. Efficacy and safety of tolvaptan versus placebo in the treatment of patients with autosomal dominant polycystic kidney disease: a metaanalysis. Int Urol Nephrol 2023;55(3):631-40.
- Hogan MC, Masyuk TV. Concurrent targeting of vasopressin receptor 2 and somatostatin receptors in autosomal dominant polycystic kidney disease: A promising approach for autosomal dominant polycystic kidney disease treatment? Clin J Am Soc Nephrol 2023;18(2):154-56.
- Sekine A, Hidaka S, Moriyama T, et al. Cystic kidney diseases that require a differential diagnosis from autosomal dominant polycystic kidney disease (ADPKD). J Clin Med 2022;11(21):6528.
- Murugapoopathy V, Gupta IR. A primer on congenital anomalies of the kidneys and urinary tracts (CAKUT). Clin J Am Soc Nephrol 2020;15(5):723-31.
- Wühl E, van Stralen KJ, Verrina E, et al. Timing and outcome of renal replacement therapy in patients with congenital malformations of the kidney and urinary tract. Clin J Am Soc Nephrol 2013;8(1):67-74.
- van der Ven AT, Vivante A, Hildebrandt F. Novel insights into the pathogenesis of monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol 2018;29(1):36-50.
- Singh S, Chaurasia A, Gopal N, et al. Treatment strategies for hereditary kidney cancer: current recommendations and updates. Discov Med 2022;34(173):205-20.
- Varshney N, Kebede AA, Owusu-Dapaah H, et al. A review of von Hippel-Lindau syndrome. J Kidney Cancer VHL 2017;4(3):20-9.
- Decks E. Belzutifan: first approval. Drugs 2021;81(16):1921-1927.
- Nair N, Chakraborty R, Mahajan Z, et al. Renal manifestations of tuberous sclerosis complex. J Kidney Cancer VHL 2020;7(3):5-19.
- Gregorio V, Caparali EB, Shojaei A, et al. Alport syndrome: clinical spectrum and therapeutic advances. Kidney Med 2023;5(5):100631.
- Аксенова М.Е. Синдром Альпорта: современные представления. Нефрология 2021;25(3):75-83 [Aksenova ME. Alport syndrome: our knowledge update. Nephrology (Saint-Petersburg) 2021;25(3):75-83 (In Russ.)].
- Dorval G, Servais A, Boyer O. The genetics of steroid-resistant nephrotic syndrome in children. Nephrol Dial Transplant 2022;37(4):648-51.
- Boyer O, Dorval G, Servais A. The genetics of steroid-resistant nephrotic syndrome in adults. Nephrol Dial Transplant 2021;36(9):1600-2.
- Lepori N, Zand L, Sethi S, et al. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin Kidney J 2018;11(2):179-90.
- Sadowski CE, Lovric S, Ashraf S et al. the SRNS Study Group. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 2015;26:1279–89.
- Santín S, Bullich G, Tazón-Vega B, et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 2011;6(5):1139-48.
- Korkmaz E, Lipska-Ziętkiewicz BS, Boyer O, et al. ADCK4-associated glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol 2016;27(1):63-8.
- Econimo L, Schaeffer C, Zeni L, et al. Autosomal dominant tubulointerstitial kidney disease: an emerging cause of genetic CKD. Kidney Int Rep 2022;7:2332-2344.
- Mabillard H, Sayer JA, Olinger E. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease. Nephrol Dial Transplant 2023;38:271-82.
- Downie ML, Lopez Garcia SC, Kleta R, Bockenhauer D. Inherited tubulopathies of the kidney: insights from genetics. Clin J Am Soc Nephrol 2021;16(4):620-30.
- Kermond R, Mallett A, McCarthy H. A clinical approach to tubulopathies in children and young adults. Pediatr Nephrol 2023;38(3):651-62.
- Foreman J. Fanconi syndrome. Pediatr Clin N Am 2019;66:159–67.
- Чеботарева Н.В., Цыгин А.Н., Буланов Н.М. и др. Синдром Фанкони у взрослых и детей. Клин фармакол тер 2022;31(1): 69-74 [Chebotareva N, Tsygin A, Bulanov N, et al. Fanconi syndrome in adults and children. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(1):69-74 (In Russ.)].
- Lemaire M. Novel Fanconi renotubular syndromes provide insights in proximal tubule pathophysiology. Am J Physiol Renal Physiol 2021;320(2):F145-60.
- Besouw MTP, Kleta R, Bockenhauer D. Bartter and Gitelman syndromes: questions of class. Pediatr Nephrol 2020;35:1815–24.
- Raina R, Krishnappa V, Das A, et al. Overview of monogenic or Mendelian forms of hypertension. Front Pediatr 2019;7:263.
- Ottosson-Laakso E, Tuomi T, Forsén B, et al. Influence of familial renal glycosuria due to mutations in the SLC5A2 gene on changes in glucose tolerance over time. PLoS One 2016;11(1):e0146114.
- Чеботарева Н.В., Цыгин А.Н., Буланов Н.М. и др. Цистиноз: патогенез, клинические проявления и лечение. Клин фармакол тер 2021;30(1):80-88. [Chebotareva N, Tsygin A, Bulanov N, et al. Cystinosis: pathogenesis, clinical features and treatment. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(1):80-8 (In Russ.)].
- Langman CB, Barshop BA, Deschênes G, et al. Controversies and research agenda in nephropathic cystinosis: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int 2016;89(6): 1192-203.
- Чеботарева Н.В., Моисеев С.В. Нефропатический цистиноз: механизмы развития и методы лечения. Клин фармакол тер 2023;32(1):79-85 [Chebotareva N, Moiseev S. Nephropathic cystinosis: pathophysiology and effects of treatment. Klinicheskaya farmakologiya i terapiya = Clin Phar macol Ther 2023;32(1):79-85 (In Russ.)].
- Emma F, van’t Hoff W, Hohenfellner K, et al. An international cohort study spanning five decades assessed outcomes of nephropathic cystinosis. Kidney Int 2021; doi.org/10.1016/j.kint.2021.06.019
- Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med 2007;147(4):242-50.
- Моисеев С.В., Тао Е.А., Моисеев А.С. и др. Клинические проявления и исходы болезни Фабри у 150 взрослых пациентов. Клин фармакол тер 2021;30(3):43-51 [Moiseev S, Tao E, Moiseev A, et al. Clinical manifestations and outcomes of Fabry disease in 150 adult patients. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(3):43-51 (In Russ.)].
- Моисеев А.С., Буланов Н.М., Тао Е.А. и др. Эффективность и безопасность длительной ферментозаместительной терапии агалсидазой альфа и агалсидазой бета у взрослых пациентов с болезнью Фабри. Клин фармакол тер 2022;31(4):28-34 [Moiseev A, Bulanov N, Tao E, et al. Efficacy and safety of long-term enzyme replacement therapy with agalsidase alfa or agalsidase beta in adult patients with Fabry disease. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(4):28-34 (In Russ.)].
- Germain DP, Elliott PM, Falissard B, et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol Genet Metab Rep 2019;19:100454.
- Лысенко (Козловская) Л.В., Рамеев В.В., Моисеев С.В. и др. Клинические рекомендации по диагностике илечению системного амилоидоза. Клин фармакол тер 2020;29(1):13-24 [Lysenko (Kozlovskaya) LV, Rameev VV, Moiseev S, et al. Clinical guidelinesfor diagnosis and treatment of systemic amyloidosis. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Therapy 2020;29(1):13-24.
- Damy T, Garcia-Pavia P, Hanna M, et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Europ J Heart Fail 2021;23:277–85
- Рамеев В.В., Моисеев С.В., Козловская Л.В. AA-амилоидоз при аутовоспалительных заболеваниях. Клин фармакол тер 2021;30(4):52-61 [Rameev V, Moiseev S, Kozlovskaya L. AA amyloidosis in autoinflammatory diseases. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(4):52-61 (In Russ.)].
- Моисеев С.В., Рамеев В.В. Дифференциальный диагноз системных аутовоспалительных заболеваний. Клин фармакол тер 2022;31(2):5-13 [Moiseev S, Rameev V. Differential diagnosis of systemic autoinflammatory diseases. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(2):5-13 (In Russ.)].
- Delaleu J, Deshayes S, Rodrigues F, et al. Tumor necrosis factor receptor-1 assciated periodic syndrome (TRAPS) related AA amyloidosis: a national case series and systematic review. Rheumatology (Oxford) 2021 Mar 14:keab252.]
- De Benedetti F, Gattorno M, Anton J, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med 2018;378:1908-19.
- Lachmann H, Kuemmerle-Deschner JB, Heike T, et al. Efficacy and safety of canakinumab in patients with cryopyrin associated periodic syndrome: results from meta-analysis of 5 studies. Arthr Rheum 2012;64(10Suppl):S321-22.
- Shee K, Stoller ML. Perspectives in primary hyperoxaluria - historical, current and future clinical interventions. Nat Rev Urol 2022;19(3):137-46.
- Groothoff JW, Metry E, Deesker L, et al. Clinical practice recommendations for primary hyperoxaluria: an expert consensus statement from ERKNet and OxalEurope. Nat Rev Nephrol 2023;19(3):194-211.
- Sas DJ, Magen D, Hayes W, et al; ILLUMINATE-B Workgroup. Phase 3 trial of lumasiran for primary hyperoxaluria type 1: A new RNAi therapeutic in infants and young children. Genet Med 2022;24(3):654-62.
- Michael M, Groothoff JW, Shasha-Lavsky H, et al. Lumasiran for advanced primary hyperoxaluria type 1: phase 3 ILLUMINATE-C trial. Am J Kidney Dis 2023;81(2):145-155.
- Коротчаева Ю.В., Козловская Н.Л., Демьянова К.А. и др. Атипичный гемолитико-уремический синдром: клиническая картина, диагностика и лечение. Клин фармакол тер 2022;31(2):43-50 [Korotchaeva Yu, Kozlovskaya N, Demyanova K, et al. Atypical hemolytic-uremic syndrome: clinical presentation, diagnosis and treatment. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022; 31(2):43-50 (In Russ.)].
- Козловская Н.Л., Прокопенко Е.И., Эмирова Х.М., Серикова С.Ю. Клинические рекомендации по диагностике и лечению атипичного гемолитикоуремического синдрома. Нефрология и диализ 2015;17(3):242-64 [Kozlov s kaya NL, Prokopenko EI, Emirova KhM, Serikova SYu. Clinical guidelines for diagnosis and treatment of atypical hemolytic uremic syndrome. Nephrology and Dialysis 2015;17(3):242-64 (In Russ.)].