Оптимальный режим дозирования препарата Ранквилон® для лечения пациентов с тревожными состояниями при неврастении и расстройствах адаптации: клиническое исследование II фазы
Тревожные расстройства — одни из наиболее распространенных психопатологических состояний, значительно ухудшающих качество жизни пациентов. Ранквилон® (АО “Валента Фарм”) продемонстрировал статистически достоверное анксиолитическое действие на различных фармакологических моделях.
Обоснование оптимального режима дозирования в отношении эффективности и безопасности препарата Ранквилон® в лечении пациентов с тревожными состояниями при неврастении и расстройствах адаптации.
В двойное слепое плацебо-контролируемое рандомизированное проспективное исследование были включены 168 взрослых пациентов с тревожными состояниями при неврастении и расстройствах приспособительного спектра, которых рандомизировали на 4 группы. Пациенты из первой группы принимали 1 таблетку препарата Ранквилон® и 2 таблетки плацебо три раза в сутки (3 мг/сут), второй – 2 таблетки препарата Ранквилон® и 1 таблетку плацебо три раза в сутки (6 мг/сут), третьей – 3 таблетки препарата Ранквилон® три раза в сутки (9 мг/сут), четвертой – 3 таблетки плацебо три раза сутки. Эффективность терапии оценивали через 28 дней на основании субшкал шкалы общего клинического впечатления (Clinical Global Impression) – улучшения (CGI-i) и тяжести (CGI-s), а также шкалы для оценки тревоги Гамильтона (HARS) и шкалы оценки астении (MFI-20). Для оценки безопасности определяли параметры жизнедеятельности, лабораторные показатели и данные электрокардиограммы (ЭКГ), а также учитывали количество и тяжесть нежелательных явлений (НЯ) и серьезных нежелательных явлений (СНЯ).
По первичному показателю эффективности (доля пациентов со значительным и выраженным улучшением по оценке врача [1 или 2 балла по шкале CGI-i] на День 29 ± 1) препарат Ранквилон® был наиболее эффективным в суточной дозе 6 мг (p=0,0148 по сравнению с плацебо).
Среди трех исследованных доз препарата Ранквилон® у пациентов с тревожными состояниями при неврастении и расстройствах адаптации оптимальной является суточная доза 6 мг, которая обеспечивает максимальные эффективность и безопасность и может быть рекомендована для изучения в клиническом исследовании III фазы.
Тревожные расстройства часто сопровождают пациентов с неврологической и соматической (кардиологической, дерматологической, онкологической, гастро интестинальной и др.) патологией. По данным Всемирной организации здравоохранения, тревожные расстройства входят в число десяти наиболее значимых проблем общественного здравоохранения. Популяци он ные исследования показывают, что на протяжении жизни с ними сталкивается от 25% до 50% населения [1]. В настоящее время тревогу рассматривают как независимый фактор риска развития неинфекционных хронических заболеваний, в том числе неврастении и расстройств адаптации [2].
Под неврастенией подразумеваются проявления астении, причиной и движущим фактором развития которой является психогенное воздействие (травмирующее переживание) [3]. По этиологическому признаку выделяют первичную (реактивную) неврастению, которая возникает на фоне стресса, эмоциональных перегрузок, переживаний, и вторичную (симптоматическую) неврастению на фоне заболеваний различных органов и систем [4]. Для неврастении характерно разнообразие клинических симптомов: астенические жалобы (общая слабость, быстрая утомляемость, сонливость, адинамия и др.), головная боль, головокружение, нарушения памяти, а также их сочетание с такими симптомами, как повышенная чувствительность к звукам, шуму и яркому свету. Важно отметить, что симптомы неврастении, как правило, не проходят после отдыха, что отличает ее от физиологической усталости [3,4].
Расстройства адаптации рассматриваются в качестве патологической реакции на эмоциональное стрессовое событие. В этих случаях стрессовый фактор не носит чрезмерный характер и не представляет непосредственной угрозы для жизни и здоровья человека. Чаще всего эмоциональное воздействие при данной патологии связано с бытовыми ситуациями, когда в силу различных причин меняется жизненный уклад или привычки, человек оказывается в условиях повышенных физических или эмоциональных нагрузок, возникают проблемы в межличностных отношениях и другие, подобные выше перечисленным, обстоятельства. В развитии дезадаптации важную роль играют индивидуальная предрасположенность к провоцирующему фактору, склонность к фиксации стрессовой ситуации и ее драматизации. Расстройства адаптации, по данным разных авторов, в популяции встречаются с частотой от 1% до 21%. В клинической картине у пациентов с данным расстройством наиболее часто отмечается тревожная или депрессивная симптоматика, а также их сочетание. Реже встречаются формы с преобладанием психовегетативных и астенических расстройств [5,6].
Развитие тревожных состояний опосредовано действием нейротрансмиттеров и биологически активных соединений, среди которых можно выделить γ-аминомасляную кислоту (ГАМК), серотонин, норадреналин, дофамин и др. [7]. Важная роль в патогенезе тревожных состояний отводится холецистокининергической системе регуляции. Холецистокинин – нейропептидный гормон, синтезируемый клетками слизистой оболочки двенадцатиперстной и тощей кишки и проксимального отдела подвздошной кишки и обладающий множеством функций, часть из которых связана с центральной нервной системой, где он оказывает влияние на процессы обучения и памяти, механизмы тревожности и болевой чувствительности (ноцицепции) [8].
Действующее вещество лекарственного препарата Ранквилон® (амид N-(6-фенилгексаноил)-глицил-Lтриптофана) является блокатором центральных холецистокининовых рецепторов. В ходе доклинических исследований было показано наличие у препарата анксиолитического эффекта, при этом он не вызывал гипноседацию, миорелаксацию, угнетение спонтанной двигательной активности и когнитивных расстройств, что выгодно отличает Ранквилон® от классических анксиолитиков [7].
Целью исследования было обоснование оптимального режима дозирования лекарственного препарата Ранквилон® в отношении эффективности и безопасности в лечении пациентов с тревожными состояниями при неврастении и расстройствах адаптации.
Материал и методы
При планировании плацебо-контролируемого исследования учитывали положения Руководств Европейского агентства по лекарственным средствам (European Medicines Agency, EMA) по клиническому изучению лекарственных препаратов, предназначенных для лечения тревожных расстройств [9,10]. Исследование было проведено в соответствии с принципами Хельсинкской декларации Всемирной медицинской ассоциации, нормативными актами Россий ской Федерации (РФ) и Евразийского экономического союза (Решение Евра зийской экономической комиссии от 3.11.2016 г. №79), а также стандартами Надлежащей Клинической Практики ICH E6 GCP. Перед проведением исследования было получено одобрение Совета по этике Минздрава РФ (№269 от 23.03.2021 г.) и разрешение Минздрава РФ (Разрешение на проведение клинических исследований №208 от 15.04.2021 г.).
В исследование включали пациентов (мужчины и женщины в возрасте от 18 до 70 лет) с тревожными расстройствами, соответствующими диагнозам неврастении (F48.0) или расстройства приспособительных реакций (F43.2) согласно Международной классификации болезней (МКБ)10. Все пациенты подписали письменное согласие на участие в данном исследовании. Критериями включения были выраженность симптомов тревоги по шкале оценки тревоги Гамильтона (Hamilton Anxiety Rating Scale – HARS) 18–24 балла, выраженность астении по шкале самооценки астении (MFI-20) более 50 баллов, сумма баллов по шкале Гамильтона для оценки депрессий (HAMD-17) менее 6, оценка по субшкале CGI-s не менее 4 баллов. До начала исследования было получено согласие пациентов на применение адекватных методов контрацепции на протяжении всего исследования и в течение 30 дней после его окончания. Для женщин с сохраненным репродуктивным потенциалом обязательным критерием включения был отрицательный тест на беременность.
Основными критериями невключения были беременность и период лактации, непереносимость действующего или вспомогательных веществ исследуемого препарата, дефицит лактазы, непереносимость лактозы или галактозы, глюкозо-галактозная мальабсорбция, прием алкоголя или наркотических препаратов на момент включения в исследование и/или алкогольная, наркотическая или лекарственная зависимость в анамнезе, онкологическая патология, инфицирование вирусом иммунодефицита человека, вирусные гепатиты, сифилис в анамнезе или в настоящее время, шизофрения, шизоаффективные, аффективные и панические расстройства, острый психоз, в том числе в анамнезе, органическое поражение центральной нервной системы травматического и алкогольного генеза, постэнцефалитический синдром, дегенеративные заболевания центральной нервной системы, депрессия и генерализованное тревожное расстройство, в том числе в анамнезе, суицидальные мысли или идеи, суицидальные попытки в анамнезе, эпилепсия, судорожные припадки, в том числе в анамнезе, а также другие тяжелые, декомпенсированные или нестабильные заболевания. В исследование не включали пациентов, которые менее чем за 90 дней до скрининга участвовали в любом другом клиническом исследовании, принимали лекарственные препараты, оказывающие выраженное влияние на гемодинамику или функцию печени (в частности, барбитураты, омепразол, циметидин) менее чем за 30 дней до начала исследования, а также пациентов, которым требовалось назначение запрещенной в рамках данного исследования сопутствующей терапии.
Исследование было проведено с 6.06.2021 г. (скрининг первого пациента) по 3.01.2022 г. (последний визит последнего пациента) на базе 8 исследовательских центров в РФ.
Исследование состояло из периода скрининга (дни от -6 до 0), периода лечения (дни от 1 до 28) с оценкой эффективности терапии на день 29±1 и периода последующего наблюдения в течение 7±1 дней. Максимальная продолжительность участия в исследовании для одного пациента составила не более 45 дней.
Пациентов рандомизировали на 4 группы: первая – 1 таблетка препарата Ранквилон® и 2 таблетки плацебо три раза в сутки (суточная доза 3 мг), вторая – 2 таблетки препарата Ранквилон® и 1 таблетка плацебо три раза в сутки (суточная доза 6 мг), третья – 3 таблетки препарата Ранквилон® три раза в сутки (суточная доза 9 мг) и четвертая – 3 таблетки плацебо три раза в сутки. Лечение продолжали в течение 28 дней.
В качестве первичной конечной точки в исследовании была принята доля больных со значительным и выраженным улучшением по оценке врача (оценка по шкале CGI-i 1 или 2 балла) на день 29±1 (Визит 3). В качестве вторичных конечных точек определяли изменения состояния пациентов по шкалам CGI-I, CGI-s, HARS, MFI-20, изменение уровня личностной и ситуативной тревожности по опроснику Спилбергера на дни 15±1 (Визит 2) и 29±1 (Визит 3), а также время достижения изменений. По данным литературы, субшкалы CGI-i и CGI-s широко используются для оценки эффективности препаратов для лечения заболеваний невротического спектра [1116].
Безопасность исследуемых доз препарата оценивали на основании мониторинга основных жизненных показателей (АД, частота сердечных сокращений и дыхательных движений), показателей общего анализа крови (концентрация гемоглобина, гематокрит, количество эритроцитов, лейкоцитов и тромбоцитов, лейкоцитарная формула, СОЭ), биохимического анализа крови (концентрации общего холестерина, общего белка, глюкозы, креатинина, общего и прямого билирубина, мочевины, активности аминотрансфераз и щелочной фосфатазы), общего анализа мочи и ЭКГ. На протяжении всего исследования оценивали количество и тяжесть НЯ и СНЯ.
Статистический анализ проводился при помощи программного обеспечения SAS® версии 9.4 (SAS Institute, Cary, NC, USA). Применяемый уровень значимости в клиническом исследовании составлял 0,05.
Результаты
Всего в исследовании было скринировано 174 пациента. Из них 168 пациентов (52 мужчины и 116 женщин) прошли скрининг и были рандомизированы на четыре группы: первая – Ранквилон® в дозе 3 мг/сут (n=43, 9 мужчин и 34 женщины, средний возраст 36,2±13,9 года), вторая – Ранквилон в дозе 6 мг/сут (n=42, 16 мужчин и 26 женщин, средний возраст 40,9±14,1 года), третья – Ранквилон® в дозе 9 мг/сут (n=41, 13 мужчин и 28 женщин, средний возраст 38,9±13,7 года) и четвертая – плацебо (n=42, 14 мужчин и 28 женщин, средний возраст 35,4±13,5 года). Досрочно выбыли из исследования 3 пациента: пропуск приема препарата – 1 пациент из первой группы, несоответствие критериям отбора – 1 пациент из второй группы, пребывание в карантине после контакта с больным COVID-19 – 1 пациентка из третьей группы (рис. 1).
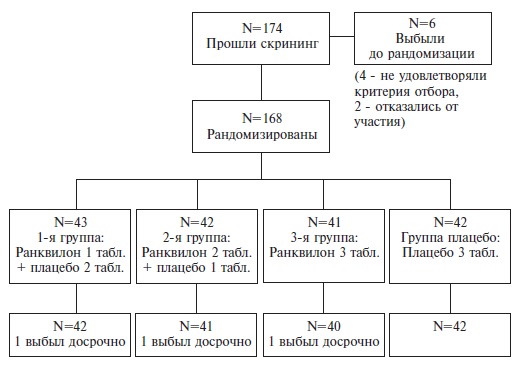
Основной анализ первичного и вторичных параметров эффективности проводился в популяции рандомизированных пациентов (intention-to-treat, ITT; n=168), дополнительный – в популяции пациентов, завершивших исследование по протоколу (per protocol, PP, n=165).
В популяции ITT значительное или выраженное улучшение по шкале CGI-i через 4 недели было достигнуто у 39,5%, 57,1% и 43,9% пациентов, получавших Ранквилон® в дозах 3, 6 и 9 мг/сут, соответственно, и 28,6% пациентов группы плацебо. В дозе 6 мг/сут препарат Ранквилон® по первичному показателю эффективности статистически значимо превосходил плацебо (p=0,0148). В популяции PP значительное или выраженное улучшение по шкале CGI-i через 4 недели было отмечено у 40,5%, 58,5% и 45,0% пациентов, получавших Ранквилон® в дозах 3, 6 и 9 мг/сут, соответственно, и 28,6% пациентов группы плацебо. В этой популяции Ранквилон® в дозе 6 мг/сут по эффективности также статистически значимо превосходил плацебо. Хотя при применении препарата в дозах 3 и 9 мг/сут частота значительного или выраженного улучшения по шкале CGI-i через 4 недели в популяциях ITT и PP была выше, чем при приеме плацебо, различия не достигли статистической значимости.
У пациентов, получавших Ранквилон® в дозе 6 мг/сут, наблюдалось более выраженное уменьшение тяжести состояния по субшкале CGI-s через 4 недели в популяции РР по сравнению с группой плацебо (р=0,0063). В этой группе было отмечено также статистически значимое улучшение суммарного балла по шкале MFI-20 через 4 недели (р=0,012) и снижение выраженности тревоги по шкале HARS в популяции ITT через 2 и 4 недели после начала терапии (р=0,0005 и р=0,0041, соответственно) по сравнению с плацебо. Кроме того, у пациентов, получавших Ранквилон® в дозе 6 мг/сут, через 4 недели значительно чаще (р=0,0204) наблюдали снижение выраженности тревоги по шкале HARS до 17 баллов и менее.
В ходе исследования не были зарегистрированы СНЯ и НЯ, которые бы привели к досрочному прекращению лечения. У 50 пациентов были отмечены 155 НЯ, среди которых 89 были расценены как связанные с приемом препарата. 47 НЯ наблюдались у 13 (30,2%) пациентов из первой группы, 25 – у 11 (26,2%) пациентов из второй группы, 35 – у 11 (26,8%) пациентов из третьей группы и 48 – у 15 (35,7%) пациентов из группы плацебо. Статистически значимых различий частоты НЯ между группами выявлено не было. Наиболее частыми НЯ были нарушения со стороны нервной системы, такие как головная боль, головокружение и сонливость, а также желудочно-кишечные нарушения (боль в животе и диарея). При оценке показателей жизнедеятельности и данных ЭКГ было зафиксировано 3 случая отклонений от нормы (1 случай повышения АД у пациента из второй группы и по 1 случаю повышения и снижения АД у пациентов из группы плацебо). Среди всех выявленных НЯ лишь одно (зубная боль у пациента из третьей группы) было расценено как тяжелое. Также были выявлены НЯ средней тяжести, в том числе 3 – у пациентов из первой группы, 2 – второй, 8 – третьей и 16 – из группы плацебо. Все остальные НЯ были легкой степени тяжести. НЯ, связанные с приемом препарата исследования, разрешились самостоятельно без последствий за исключением одного случая протеинурии (2,3%) в первой группе, исход которой остался неизвестным. Статистически значимых различий при оценке безопасности между группами приема препарата и плацебо не выявлено.
Обсуждение
Клинические исследования II фазы являются важной и неотъемлемой частью разработки оригинальных лекарственных препаратов. Они позволяют определить, обладает ли новый препарат или метод лечения достаточной эффективностью и безопасностью, чтобы гарантировать успех его дальнейшего изучения в крупномасштабном рандомизированном клиническом исследовании III фазы. Исследования II фазы также дают представление о спектре заболеваний, при которых препарат наиболее эффективен, возможных НЯ и их лечении, а также об оптимальных режимах дозирования для планирования последующих исследований. По данным Управления по контролю за качеством пищевых продуктов и лекарственных средств (FDA), примерно 33% препаратов, изучающихся в исследованиях II фазы, переходят в следующую фазу клинических исследований [17,18].
Несмотря на многообразие возможных дизайнов исследований фазы II "золотым стандартом" для доказательства эффективности нового препарата в таких исследованиях и перехода к проведению регистрационных исследований III фазы являются плацебо-контролируемые рандомизированные исследования [17]. В исследованиях II фазы обычно участвуют до нескольких сотен пациентов, а продолжительность лечения варьируется от нескольких месяцев до нескольких лет [18].
Стратегия подбора доз и выбора конечных точек в клиническом исследовании II фазы напрямую влияет на результаты оценки общей эффективности препарата в клиническом исследовании III фазы. Выбор одной или нескольких доз для перехода к III фазе клинических исследований является одним из наиболее сложных решений при разработке лекарственных средств. Отсутствие успеха клинического исследования III фазы часто можно объяснить неправильным подбором дозы, которая слишком мала для достижения желаемого эффекта или слишком высока, что приводит к неблагоприятному профилю безопасности. Правильно подобранная доза и схема дозирования будут иметь более благоприятное соотношение польза/риск, что в конечном итоге напрямую определяет перспективность новой лечебной технологии [19].
Все вышеизложенные принципы были учтены при планировании клинического исследования II фазы препарата Ранквилон, которое было двойным слепым, рандомизированным, плацебо-контролируемым и предполагало изучение эффективности и безопасности суточных доз 3 мг, 6 мг и 9 мг. Одной из целей исследовании была оценка наличия взаимосвязи "дозаэффект". При планировании исследования были учтены требования руководств по клиническому изучению лекарственных препаратов, предназначенных для лечения тревожных расстройств.
Полученные результаты позволили оценить безопасность и эффективность препарата Ранквилон®, а также проанализировать данные о возможной взаимосвязи "доза-эффект". Частота НЯ при различных режимах дозирования Ранквилона® существенно не отличалась. Полученные данные показали эффективность препарата Ранквилон® в лечении пациентов с тревожными состояниями при неврастении и расстройствах адаптации. Наибольший эффект по сравнению с плацебо при сопоставимой безопасности был достигнут при приеме препарата Ранквилон® в суточной дозе 6 мг.
Заключение
Результаты исследования II фазы свидетельствуют о том, что оптимальная доза препарата Ранквилон® у пациентов с тревожными состояними при неврастении и расстройствах адаптации составляет 6 мг/сут. Эффективность данной дозы препарата превосходила плацебо при сопоставимом профиле безопасности. Наиболее часто встречавшимися НЯ, вероятно, связанными с использованием препарата Ранквилон®, были нарушения со стороны нервной системы (головная боль, головокружение и сомнолентность) и желудочнокишечные нарушения (боль в животе и диарея). Доза препарата Ранквилон® 6 мг может быть рекомендована для изучения в клиническом исследовании III фазы.
Используемые источники
- Гуров В.А., Медведев В.Э. Тревожные расстройства в общей врачебной практике: аспекты клиники и терапии. Архивъ внутренней медицины 2011;2:15–9 [Gurov VA, Medvedev VE. Anxiety disorders in general medical practice: aspects of the clinic and therapy. The Russian Archives of Internal Medicine 2011;2:15–9 (In Russ.)].
- Шишкова В.Н., Драницына Б.Г., Драпкина О.М. Алгоритмы ведения пациентов с тревогой в практике терапевта. Кардиоваскулярная терапия и профилактика 2023;22(2):63–9 [Shishkova VN, Dranitsyna BG, Drapkina OM. Algorithms for the management of patients with anxiety in the internist’s practice. Cardiovascular Therapy and Prevention 2023;22(2):63-9 (In Russ.)].
- Чутко Л.С., Баранова И.А. Неврастения. Медицинский совет 2009;4:23–4 [Chutko LS, Baranova IA. Neurasthenia. Medical Council (Meditsinskiy sovet) 2009;(4):23–4 (In Russ.)].
- Дадашева К.Н., Агафонов Б.В., Дадашева М.Н., Подрезова Л.А. Неврастенический синдром в общеврачебной практике. Возможности терапии. РМЖ. Медицинское обозрение 2019;4(II):91–5 [Dadasheva KN, Agafonov BV, Dadasheva MN, Podrezova LA. Asthenic syndrome in general practice: therapy possibilities. RMJ. Medical Review 2019;4(II):91–5 (In Russ.)].
- Антипова О.С. Расстройства адаптации: современные походы к диагностике и терапии. Психиатрия и психофармакотерапия 2012;14(6):22–7 [Antipova OS. Adaptation disorders: modern approaches to diagnosis and therapy. Psychiatry and psychopharmacotherapy 2012;14(6):22–7 (In Russ.)].
- Пацкань И.И. Современная клинико-эпидемиологическая характеристика медико-социальной проблемы расстройств адаптации. Университетская клиника 2019;3(32):87–92 [Patskan II. Modern clinical and epidemiological characteristics of the medical and social problem of adjustment disorders. University Clinic 2019;3(32):87–92 (In Russ.)].
- Незнамов Г.Г., Дорофеева О.А., Метлина М.В. и др. Результаты клинического исследования нового анксиолитика, блокатора центральных холецистокининовых рецепторов. Журнал неврологии и психиатрии им. С.С. Корсакова 2019;119(8):53-60 [Neznamov GG, Dorofeeva OA, Metlina MV, t al. Results of a clinical study of a new anxiolytic, a blocker of central cholecystokinin receptors. S.S. Korsakov Journal of Neurology and Psychiatry = Zhurnal Nevrologii i Psikhiatrii im. S.S. Korsakova 2019;119(8):53-60 (In Russ.)].
- Chandra R, Liddle RA. Cholecystokinin. Curr Opin Endocrinol Diab Obesity 2007;14(1):63-7.
- European Medicines Agency. Guideline on the clinical investigation of medicinal products indicated for generalised anxiety disorder. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinicalinvestigation-medicinal-products-indicated-generalised-anxiety-disorder_en.pdf.
- European Medicines Agency. Guideline on clinical investigation of medicinal products indicated for the treatment of panic disorder. London, 20 January 2005.
- Зозуля А.А., Незнамов Г.Г., Сюняков Т.С. и др. Эффективность и механизмы действия нового дипептидного анксиолитика Селанка при терапии генерализованного тревожного расстройства и неврастении. Журнал неврологии и психиатрии им С.С. Корсакова 2008;108(4):38–49 [Zozulya AA, Neznamov GG, Syunyakov TS, et al. Efficacy and possible mechanism of action of a new peptide anxiolytic Selank in the therapy of generalized anxiety disorders and neurasthenia. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova 2008;108(4):38–49 (In Russ.)].
- Медведев В.Э., Терещенко О.Н., Кост Н.В.и др. Оптимизация терапии тревожных расстройств пептидным препаратом Селанк. Журнал неврологии и психиатрии им С.С. Корсакова 2015;115(6):33-40 [Medvedev VE, Tereshechenko ON, Kost NV, et al. Optimization of the treatment of anxiety disorders with selank. Zhurnal Nevrologii i Psikhiatrii imeni S.S. Korsakova 2015;115(6):33-40 (In Russ.)].
- Медведев В.Э., Епифанов А.В. Терапия невротических, связанных со стрессом, и соматоформных расстройств у пациентов с гипертонической болезнью. Российский психиатрический журнал 2011;(1):55–61 [Medvedev VE, Epifanov AV. Therapy of neurotic, stress-related and somatoform disorders in hypertonic patients. Russian Journal of Psychiatry 2011;1:55–61 (In Russ.)].
- Сюняков Т. С., Незнамов Г. Г. Оценка терапевтической эффективности и безопасности селективного анксиолитика афобазола при генерализованном тревожном расстройстве и расстройствах адаптации: результаты многоцентрового рандомизированного сравнительного с диазепамом исследования. Терапевтический архив 2016;88(8):73-86 [Syunyakov TS, Neznamov GG. Evaluation of the therapeutic efficacy and safety of the selective anxiolytic afobazole in generalized anxiety disorder and adjustment disorders: Results of a multicenter randomized comparative study of diazepam. Terapevticheskii Arkhiv 2016;88(8):73-86 (In Russ.)].
- Яхно Н.Н., Парфенов В.А., Рейхарт Д.В. и др. Многоцентровая неинтервенционная проспективная наблюдательная программа изучения практики назначения препарата тералиджен у больных с диагнозом вегетативного расстройства (СТАРТ-2: российский опыт применения русскоязычной валидированной версии опросника 4DSQ. Промежуточный анализ). Журнал неврологии и психиатрии 2015;115(5):27–33 [Yakhno NN, Parfenov VA, Reyhart DV, et al. The multicenter non-interventional, prospective observational program on the study of practical use of teraligen in patients diagnosed with autonomic disorder (START2): a local Russian experience with the use of the Russian version of The Four-Dimensional Symptom Questionnaire (4DSQ). Zhurnal Nevrologii i Psikhiatrii 2015;115(5):27–33 (In Russ.)].
- Смулевич А.Б., Андрющенко А.В., Бескова Д.А. Новый подход к терапии неврастении и соматогенной астении (результаты многоцентрового исследования эффективности и безопасности Ладастена). Психиатрия и психофармакотерапия 2009;11(1):18–26 [Smulevich АВ, Andryushchenko AV, Bes kova DA. A new approach to the treatment of neurasthenia and somatogenic asthenia (results of a multicenter study of the efficacy and safety of Ladasten). Psychiatry and psychopharmacotherapy 2009;11(1):18–26 (In Russ.)].
- Torres-Saavedra PA, Winter KA. An overview of phase 2 clinical trial designs. Int J Radiat Oncol Biol Phys 2022;112(1):22-9.
- U.S. Food and Drug Administration (FDA). Step 3: Clinical Research. The Drug Development Process; https://www.fda.gov/patients/drug-development-process/step-3-clinical-research.
- Antonijevic Z, Pinheiro J, Fardipour PF, Lewis RJ. Impact of dose selection strategies used in phase II on the probability of success in phase III. Stat Biopharm Res 2010;2(4):469-86.