Биоаналоги в лечении ревматических болезней: pro et contra
Генно-инженерные биологические препараты (ГИБП) в настоящее время достаточно широко применяются для лечения многих ревматических болезней, в том числе ревматоидного артрита, анкилозирующего спондилита, псориатического артрита, системных васкулитов и др. Чаще всего их используют в качестве препаратов второй линии в случае неэффективности или плохой переносимости стандартных базисных противовоспалительных препаратов. Основным препятствием к более частому назначению ГИБП является их высокая стоимость, которая приводит к увеличению затрат на лечение и экономического бремени для системы здравоохранения. Частичным решением этой проблемы является создание экономически более привлекательных биоаналогов ГИБП, которое стало возможным после завершения срока патентов на некоторые оригинальные препараты. Однако следует учитывать, что ГИБП, в отличие от синтетических лекарственных средств (“малых молекул”), характеризуются сложным и многоэтапным процессом производства, что определяет строгие требования к их разработке и регистрации. Чтобы гарантировать не менее высокую эффективность и безопасность биоподобных препаратов по сравнению с оригинальными, потенциальные биоаналоги следует тщательно изучать не только в доклинических, но и рандомизированных контролируемых клинических исследованиях. Сбор данных о безопасности и эффективности биоаналога необходимо продолжать и после его регистрации и внедрения в клиническую практику. В статье обсуждаются результаты клинических исследований российских биоаналогов ритуксимаба, инфликсимаба и адалимумаба, которые были одобрены для применения при ревматических заболеваниях.
Генно-инженерные биологические препараты (ГИБП) применяются в ревматологии на протяжении двух десятилетий. За это время список их значительно пополнился и продолжает расширяться (ингибиторы фактора некроза опухоли [ФНО]-α, В-лимфоцитов, рецепторов интерлейкина-6, костимуляции Т-лимфоцитов, интерлей кинов-12/23, интерлейкина-17, интерлейкина-1), как и показания к применению. Последние сегодня включают не только ревматоидный артрит, но и многие другие ревматические заболевания, в том числе редкие. К настоящему времени у некоторых ГИБП закончился срок патентной защиты, в связи с чем начался процесс регистрации их аналогов. На сегодняшний день в Российской Федерации для лечения ревматических заболеваний были одобрены биоаналоги инфликсимаба (инфликсимаб, BIOCAD, Россия, и Фламмэгис, Celltrion Pharm, Республика Корея), ритуксимаба (Ацеллбия, BIOCAD, Россия) и адалимумаба (Далибра, BIOCAD, Россия.)
Современные принципы разработки биоаналогов
Появление более дешевых биоаналогов приводит к снижению затрат для системы здравоохранения, в том числе за счет падения цен на оригинальные ГИБП, что позволяет охватить лечением большее число пациентов. Например, в одном исследовании было показано, что появление биоаналогов на американском рынке в течение ближайшего десятилетия позволит сократить расходы бюджета более чем на 44 млрд долларов при условии, что стоимость биоподобных препаратов будет на 35% ниже оригинальных ГИБП [1]. Однако очевидно, что сокращение затрат не должно происходить в ущерб эффективности и безопасности лечения ГИБП. В связи с этим в литературе последних лет активно обсуждаются требования к процессу регистрации биоаналогов, в частности необходимость в проведении рандомизированных клинических исследований для подтверждения их сопоставимых эффективности и безопасности по сравнению с оригинальным ГИБП [2-5]. Если подобные исследования не проводились, то препарат предлагается считать “предполагаемой копией" (intended copy) референтного ГИБП, а не биоаналогом.
ГИБП, применяющиеся для лечения ревматических заболеваний, представляют собой сложные белковые молекулы (моноклональные антитела, циркулирующие рецепторы, гибридные молекулы), которые производятся с помощью генно-инженерного метода и, в отличие от низкомолекулярных препаратов, не являются точными копиями оригинальных веществ. Это связано с тем, что даже незначительные изменения процесса производства белковых молекул могут привести к изменению их биологической активности и/или иммуногенности и, соответственно, безопасности и эффективности [3]. Свойства белков могут зависеть и от трудно воспроизводимых посттрансляционных изменений, таких как гликозилирование, окисление, метилирование и/или деаминирование [5].
В последние годы Всемирная организация здравоохранения (ВОЗ) и регуляторные органы Европейского Союза и США предложили определения биоаналогов. Например, ВОЗ считает таковыми биотерапевтические препараты, которые по качеству, безопасности и эффективности подобны зарегистрированным референтным препаратам [6]. В определении Европейского агентства по лекарствам (ЕМА) и сходном определении Евразийской экономической комиссии указано, что подобие биоаналога референтному препарату по параметрам качества, биологической активности, безопасности и эффективности должно быть подтверждено при всестороннем исследовании сопоставимости [7,8]. В определении Американского управления по контролю качества пищевых продуктов и лекарств (FDA) отмечается возможность незначительных различий в клинически неактивных компонентах между биоаналогом и зарегистрированным в США оригинальным ГИБП при отсутствии клинически значимых различий по безопасности, чистоте и активности [9].
Сегодня большинство экспертов сходятся во мнении, что регистрация биоаналога должна производиться на основании результатов не только доклинических, но и клинических исследований 3 фазы, предполагающих прямое сравнение потенциального биоаналога с оригинальным ГИБП (рис. 1) [2,5]. Таким образом, фактически программа разработки биоаналога в значительной степени соответствует таковой оригинального препарата за исключением отсутствия необходимости в проведении исследований 2 фазы, предполагающих изучение режимов введения и выбор оптимальных доз препарата. Регуляторные органы Европы и США, а также Евразийская экономическая комиссия считают целесообразным придерживаться пошагового подхода при изучении биоаналогов [7-9]. При этом объем и характер доклинических и клинических исследований зависят от результатов, полученных на предыдущем этапе. Следует отметить, что необходимо учитывать все полученные данные, а не отдавать предпочтение тому или иному аспекту. Цель любых исследований – выявление потенциальных различий между биоаналогом и оригинальным ГИБП и установление клинического значения различий, если, конечно, таковые будут обнаружены [2].
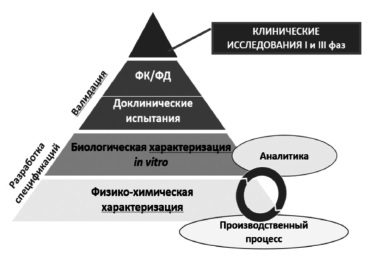
В доклинических исследованиях изучают аминокислотную последовательность, физико-химические свойства, показатели качества белков, подтверждают сопоставимость серий биоаналога и оригинального ГИБП по различным характеристикам, таким как наличие заряженных изоформ, гликозилирование и другие посттрансляционные изменения, составу примесей [8]. Ключевое значение имеет сравнение функциональных свойств ГИБП, таких как связывание моноклональных антител с антигеном-мишенью или Fc-рецепторами, антителозависимая клеточная и комплементзависимая цитотоксичность, активация комплемента [8]. Методы анализа должны быть достаточно чувствительными для выявления различий зависимости активности от концентрации между биоподобным и референтным препаратами. Это означает, что в доклинических исследованиях важно не просто показать наличие у белка определенного свойства, а доказать, что по соответствующему показателю он не отличается от оригинального ГИБП, т.е. является биоподобным.
Такая же цель преследуется в клинических исследованиях, в которых изучают фармакокинетическую и фармакодинамическую эквивалентность биоаналога и оригинального ГИБП (границы сопоставимости по первичным параметрам обычно составляют 80-125%) и подтверждают их сопоставимые клиническую эффективность и безопасность.
Как указано выше, необходимым этапом разработки биоаналога является подтверждение его сопоставимости с оригинальным ГИБП в рандомизированном контролируемом исследовании 3 фазы, которое следует проводить в наиболее “чувствительной" выборке пациентов [2]. Такие исследования должны быть достаточно продолжительными, чтобы изучить сохранение достигнутого ответа в отдаленные сроки и безопасность и иммуногенность более длительной терапии. Одно вре менно сопоставление результатов лечения в более ранние сроки, например, через 8-12 недель, позволяет сравнить скорость действия препаратов сравнения [10]. Выбор критериев эффективности в сравнительных клинических исследованиях должен быть научно обоснованным. При оценке эффективности необходимо свести к минимуму роль факторов, которые зависят от пациента и заболевания.
Любые белки обладают иммуногенностью, т.е. могут вызывать образование антител, в том числе нейтрализующих, которые нивелируют эффективность лечения ГИБП. По иммуногенности биоаналоги могут гипотетически отличаться от оригинальных препаратов, особенно если в процессе их производства используются иные экспрессирующие конструкции, которые могут привести к модификации свойств биологического препарата [8]. Соответственно, при проведении сравнительного клинического исследования необходимо определять общую частоту образования антител и частоту образования как связывающих, так и особенно нейтрализующих антител, а также их влияние на эффективность ГИБП. Изучение иммуногенности более информативно у пациентов, которым ранее не проводилась терапия ГИБП [11]. В идеале они также не должны получать сопутствующую терапию иммуносупрессивными препаратами. Необходимо учитывать, что иммуногенность ГИБП может зависеть от дозы ГИБП, генетических факторов, особенностей заболевания [2]. Например, в сравнительных клинических исследованиях оригинального инфликсимаба и его биоаналога частота антител к инфликсимабу у пациентов с ревматоидным артритом, получавших инфликсимаб в дозе 3 мг/кг и метотрексат, была выше, чем у больных анкилозирующим спондилитом, которым проводили терапию одним инфликсимабом в дозе 5 мг/кг [12,13].
Экстраполяция показаний к применению биоаналого
Нужно ли подтверждать биоподобие биоаналога и референтного препарата по каждому показанию к применению? Некоторые специалисты считают необходимым проводить рандомизированные клинические исследования по каждому зарегистрированному показанию [14,15]. Однако подобная практика представляется чрезмерной. По мнению группы экспертов, если на этапе доклинических исследований доказана эквивалентность качественных и функциональных характеристик, а в рандомизированном клиническом исследовании подтверждена сопоставимость клинической эффективности и безопасности биоаналога оригинальному препарату по крайней мере по одному показанию, то полученные данные могут быть экстраполированы на другие показания, по которым биоаналог не изучался [2]. Данная практика правомочна в отношении тех показаний, при которых механизм действия одинаков. Так, многие ГИБП в ревматологии применяются по различным показаниям, например, ингибиторы ФНО-α используют для лечения ревматоидного артрита, анкилозирующего спондилита, псориатического артрита, а также псориаза и воспалительных заболеваний кишечника. Cпециально изучать фармакокинетические свойства или клиническую эффективность биоаналогов ингибиторов ФНО-α у пациентов со всеми заболеваниями, которые служат показаниями к их применению, не имеет особого смысла, учитывая однотипную роль ФНО-α в иммуновоспалительном процессе [2]. Однако некоторые ГИБП, например, ритуксимаб, используются у пациентов не только с ревматическими, но и онкологическими заболеваниями, что может потребовать проведения отдельных фармакокинетических исследований, учитывая потенциальные различия мишень-опосредованного клиренса.
В настоящее время экстраполяция показаний для биоаналогов стала общемировой практикой, которая позволяет производителю не только сократить затраты на клинические исследования и обеспечить препарату более привлекательную стоимость, но и быстрее внедрить его в клиническую практику.
Переключение пациентов с оригинального препарата на биоаналог
Применение биоаналогов в качестве первого ГИБП получает все большее распространение, так как подобная практика увеличивает доступность современных методов лечения для пациентов и ограничивает рост затрат системы здравоохранения. Однако для практикующего специалиста остро стоит вопрос о возможности переключения пациентов с оригинальных ГИБП на биоаналоги. Этот вопрос изучался в клинических исследованиях [16-19] и затрагивается в последних международных рекомендациях по ревматоидному артриту [20] и анкилозирующему спондилиту [21]. В клинической практике можно встретить следующие ситуации, связанные с переключениями между референтным препаратом и его биоаналогом.
- Оригинальный препарат эффективен, однако для уменьшения финансовых затрат учреждения/области/ региона может быть рекомендовано переключение на биоаналог с аналогичным международным непатентованным названием (МНН). Данное переключение, как правило, вызывает наименьшее количество вопросов и у клиницистов, и у регуляторов [2]. Более того, по мнению группы экспертов нет оснований ожидать снижение эффективности лечения при переключении между двумя биоаналогами [2]. Недавно Норвежское агентство по лекарствам (Norwegian Medicines Agency) предложило разрешить автоматическую замену оригинальных препаратов на биоаналоги на уровне аптеки [22]. При этом важно предоставить пациенту адекватную информацию о качестве и изученности предлагаемого биоаналога, чтобы избежать эффекта ноцебо и “снижения" эффективности терапии, которое может быть связано с недоверием к новому препарату. Кроме того, остается открытым вопрос иммуногенности при множественных переключениях между биоаналогами и оригинальным препаратом или между биоаналогами в рамках одного МНН.
- Оригинальный препарат эффективен, однако для уменьшения финансовых затрат учреждения/области/ региона может быть рекомендовано переключение на биоаналог с другим МНН. Данное переключение требует веского обоснования, так как смена МНН даже внутри одного класса (например, среди ингибиторов ФНО-α) может привести к потере клинического эффекта.
- Оригинальный препарат неэффективен и обсуждается переключение на биоаналог. В подобной ситуации очевидно, что переключение следует проводить на биоподобный препарат с другим МНН, так как продолжение лечения неэффективным препаратом клинически нецелесообразно [20,21]. К сожалению, к настоящему времени накоплено недостаточно информации об оптимальных схемах переключения даже между оригинальными ГИБП, и тем более почти совсем нет опыта переключения с референтных препаратов на биоаналоги с другими МНН.
Российская практика разработки и применения биоаналогов
С 2017 г. в Российской Федерации по ревматическим показаниям зарегистрировано 3 биоаналога ГИБП российского производства: ритуксимаб (Ацеллбия, BIOCAD), инфликсимаб (Инфликсимаб, BIOCAD) и адалимумаб (Далибра, BIOCAD). Разработка и полный цикл локального производства российских биоаналогов позволили выпустить на рынок препараты высокого качества по более конкурентной цене. Ниже мы приводим краткий обзор клинических исследований данных биоподобных препаратов.
Биоаналог ритуксимаба (Ацеллбия®). Регистрационное исследование препарата Ацеллбия® (BCD-020, МНН: ритуксимаб) – BIORA – проводилось у пациентов с серопозитивным ревматоидным артритом на базе 21 аккредитованного лечебного центра в России, Беларуси, Украине и Индии [23,24]. Эффективность терапии оценивали у 160 пациентов, 83 из которых получали биоаналог, а 77 – референтный ритуксимаб (Мабтера®). Пациенты двух групп были сопоставимы по возрасту, антропометрическим показателям, продолжительности (в среднем около 7 лет) и активности заболевания. Для оценки эффективности терапии исполь зовали критерии Американской коллегии ревматологов (ACR)/Европейской антиревматической лиги (EULAR) и критерии ACR (ACR20/50/70). Исследо вание проводилось в два этапа: первый – оценка биоподобия (24 недели), второй – оценка взаимозаменяемости (24 недели).
Через 24 недели после начала лечения частота улучшения по критериям ACR20 в группе пациентов, получавших биоаналог ритуксимаба, составила 84,1% (95% доверительный интервал [ДИ] 74,8–90,5), а в группе сравнения – 87,0% (95% ДИ 77,7–92,8). Разница между группами была недостоверной (р=0,773) [23]. На втором этапе исследования, когда изучались результаты переключения пациентов, ранее получавших лечение референтным препаратом, на биоаналог, и наоборот, не было отмечено изменений эффективности, безопасности и иммуногенности терапии.
В Российской Федерации зарегистрированные показания к применению референтного ритуксимаба и его биоаналога в ревматологии включают в себя не только ревматоидный артрит, но и АНЦА-ассоциированные васкулиты – гранулематоз с полиангиитом и микроскопический полиангиит. Регистрационное исследование препарата Ацеллбия у пациентов с АНЦА-ассоциированными васкулитами не проводилось, т.е. это показание было зарегистрировано на основании опыта изучения оригинального ГИБП и принципа экстраполяции. Следует отметить, что эффективность и безопасность Ацеллбии были показаны в ретроспективном исследовании у 42 больных АНЦА-ассоциированным васкулитом, выполненном на базе Клиники им. Е.М. Тареева (Москва) [25].
Биоаналог инфликсимаба (Инфликсимаб). Российский биоаналог Инфликсимаб (BCD-055) был зарегистрирован в Российской Федерации на основании результатов двух клинических исследований 3 фазы: ASART-2 (анкилозирующий спондилит) и LIRA (ревматоидный артрит). Биоаналог сравнивали с референтным препаратом Ремикейд® (“МСД Фармасьютикалс").
В международное многоцентровое, рандомизированное, двойное слепое клиническое исследование ASART2 были включены 199 пациентов с анкилозирующим спондилитом, которых рандомизировали на две группы в соотношении 2:1. Им назначали BCD-055 или Ремикейд в дозе 5 мг/кг в режиме 0–2–6-я неделя, затем каждую 8-ю неделю. Результаты оценивали через 14, 30 и 54 недель у всех рандомизированных пациентов, получивших хотя бы одну дозу инфликсимаба (выборка intent-to-treat; ITT), и через 54 недели у пациентов, завершивших участие в исследовании согласно протоколу (выборка per protocol; РР). Частота ответа по критериям ASAS20 и ASAS40 через 14, 30 и 54 недели была сопоставимой в группах сравнения (р>0,05) [26]. Через 54 недели доля пациентов, достигших ответа по критериям ASAS20 при лечении BCD-055 и Реми кей дом, в выборке ITT составила 67,4% и 52,2%, соответственно (р=0,053), а в выборке РР – 80,9% и 68,6% (р=0,128). Частота ответа по критериям ASAS40 в двух группах составила, соответственно, 53,0% и 38,8% в выборке ITT (р=0,081) и 63,6% и 50,9% в выборке РР (р=0,177) [26].
В многоцентровом исследовании LIRA у 426 пациентов с активным ревматоидным артритом, рандомизированных на две группы в соотношении 2:1 (BCD-055 и Ремикейд), биоаналог инфликсимаба не отличался от референтного ГИБП по частоте ответа по критериям ACR20/50/70 и частоте ремиссии по критерию ACR/ EULAR [27].
Биоаналог адалимумаба (Далибра®). В 2019 году был зарегистрирован российский биоаналог адалимумаба (Далибра®). Регистрационное исследование 3 фазы проводилось у 346 пациентов со средне-тяжелым и тяжелым псориазом, которые были рандомизированы на две группы в соотношении 1:1. В этом исследовании была установлена эквивалентность исследуемого препарата BCD-057 (Далибра®, BIOCAD) оригинальному адалимумабу (Хумира®, Веттер Фарма-Фертигунг Гмбх и Ко.КГ, Германия) по эффективности, безопасности, фармакокинетике и иммуногенности. Также продемонстрировано отсутствие негативного влияния на описанные параметры переключения с оригинального препарата на биоаналог [28]. Весь спектр показаний к применению оригинального препарата был экстраполирован на биоаналог.
Заключение
Выход на рынок биоаналогов и их внедрение в клиническую практику позволяет существенно снизить затраты системы здравоохранения и обеспечить большее число пациентов эффективной терапией. Как правило, в течение 2-3 лет после выхода на рынок биоаналогов наблюдаются следующие тенденции: (1) снижение рыночной стоимости соответствующих МНН; (2) повышение доступности терапии для пациентов без дополнительной нагрузки на государственный бюджет; (3) замещение оригинальных препаратов в государственных закупках биоаналогами. В связи с увеличением частоты использования биоаналогов перед регуляторами встает вопрос о возможной взаимозаменяемости оригинальных препаратов и их копий. В Европейском союзе полномочия по решению этого вопроса переданы на национальный уровень. В настоящее время принято считать, что переключение с референтного препарата на его биоаналог является безопасным и эффективным. Более того, нет оснований предполагать другой кли нический исход при переключении между двумя биоаналогами. Обоснованность данного подхода подтверждается анализом опыта реальной клинической практики и национальных регистров.
Используемые источники
- Mulcahy AW, Predmore Z, Mattke S. The cost savings potential of biosimilar drugs in the United States. Perspectives. 2014 http://www. rand. org/ pubs/ perspectives/ PE127.html (accessed 6 Nov 2019).
- Kay J, Schoels MM, Dörner T, et al. Consensus-based recommendations for the use of biosimilars to treat rheumatological diseases. Ann Rheum Dis 2018;77(2):165-74.
- Зырянов С.К., Белоусов Ю.Б. Биоаналоги в современном здравоохранении: что нужно знать клиницисту? Клин фармакол тер 2011;1:18-20 [Zyryanov SK, Belousov YuB. Biosimilars in the modern health care system: what should clinician know? Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2011;1:18-20 (In Russ.)].
- Новиков П.И., Моисеев С.В. Биоаналогичные лекарственные препараты – перспективы использования в ревматологии. Клин фармакол тер 2015; 24(1):13-7 [Novikov PI, Moiseev SV. The perspectives on biosimilars use in rheumatology. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2015;24(1):13-7 (In Russ.)].
- Моисеев С.В., Новиков П.И. Роль биоподобных лекарственных препаратов в лечении ревматических заболеваний. Клин фармакол тер 2018;27(5):17-22 [Moiseev SV, Novikov PI. The role of biosimilars in the treatment of rheumatic diseases. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2018; 27(5):17-22 (In Russ.)].
- World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). http://www.who.int/biologicals/areas/biological_ therapeutics/ BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed 28 March 2018.
- European Medicines Agency. Guideline on similar biological medicinal products (Revision). CHMP/437/04 Rev 1. London, UK: 2014.
- Евразийская экономическая комиссия. О правилах регистрации и экспертизы лекарственных средств для медицинского применения. Решение 03.11.2016. https://docs.eaeunion.org/docs/ru-ru/01411969/cncd_21112016_78. [Eurasian Economic Comission. The rules of drugs approval and evaluation. 03.11.2016. https://docs.eaeunion.org/docs/ru-ru/01411969/cncd_21112016_78. (In Russ.)].
- US Department of Health and Human Services, US Food and Drug
- Administration. Guidance for industry scientific considerations in demonstrating biosimilarity to a reference product. Rockville, MD:US Dept of Health and Human Services; 2015.
- Kay J, Smolen JS. Biosimilars to treat inflammatory arthritis: the challenge of proving identity. Ann Rheum Dis 2013;72:1589–93.
- Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014 http://www. ema. europa. eu/ docs/ en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf (accessed 6 Nov 2019).
- Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis 2013;72:1605–12.
- Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis 2013;72: 1613–20.
- Danese S, Fiorino G, Michetti P. Viewpoint: knowledge and viewpoints on biosimilar monoclonal antibodies among members of the European Crohn’s and Colitis Organization. J Crohns Colitis 2014;8:1548–50.
- Fiorino G, Danese S. The biosimilar road in inflammatory bowel disease: the right way? Best Pract Res Clin Gastroenterol 2014;28:465–71.
- Park W, Yoo DH, Miranda P, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis 2017;76:346–54.
- Emery P, Vencovský J, Sylwestrzak A, et al. Long-term efficacy and safety in patients with rheumatoid arthritis continuing on SB4 or switching from reference etanercept to SB4. Ann Rheum Dis 2017;76:1986–91.
- Jørgensen KK, Olsen IC, Goll GL, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet 2017;389:2304–16.
- Numan S, Faccin F. Non-medical switching from originator tumor necrosis factor inhibitors to their biosimilars: systematic review of randomized controlled trials and real-world studies. Adv Ther 2018;35:1295–332.
- Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960–977.
- Ward M, et al 2019 Update of the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network Recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthr Care Res 2019; Published Online First. https://legemiddelverket.no/nyheter/switching-between-a-reference-product-anda-biosimilar.
- Насонов Е.Л., Зонова Е.В., Иванова О.Н. и др. Результаты сравнительного клинического исследования III фазы препаратов ритуксимаба (Ацеллбия и Мабтера) при ревматоидном артрите (исследование BIORA). Научно-практическая ревматология 2016;54(5):510-9 [Nasonov EL, Zonova EV, Ivanova ON, et al. The results of a phase III comparative clinical trial of rituximab (Acellbia and MabThera) in rheumatoid arthritis (the BIORA study). NauchnoPrakticheskaya Revmatologiya = Rheumatology Science and Practice. 2016;54(5): 510-9 (In Russ.)].
- Каратеев ДЕ, Мазуров ВИ, Зонова ЕВ и др. Сравнительная эффективность и безопасность биоаналога инфликсимаба (BCD-055) и оригинального инфликсимаба у пациентов с анкилозирующим спондилитом (результаты международных многоцентровых рандомизированных двойных слепых клинических исследований I и III фазы). Современная ревматология. 2017; 11(3):14–25 [Karateev DE, Mazurov VI, Zonova EV, et al. Comparative efficacy and safety of infliximab biosimilar (BCD-055) and innovator infliximab in patients with ankylosing spondylitis (results of international, multiple-center, double-blind phase I and phase III clinical studies). Sovremennaya Revmatologiya = Modern Rheumatology Journal 2017;11(3):14–25 (In Russ.)].
- Новиков П.И., Зыкова А.С., Щеголева Е.М. и др. Оценка краткосрочной эффективности и безопасности биоаналога ритуксимаба при АНЦА-ассоциированных васкулитах. Клин фармакол тер 2018;27(2):38-42 [Novikov PI, Zykova AS, Shchegoleva EM, et al. Short-term efficacy and safety of biosimilar rituximab in ANCA-associated vasculitis. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2015;24(1):13-7 (In Russ.)].
- Лила А.М., Мазуров В.И., Зонова Е.В. и др. Сравнительная оценка долгосрочной эффективности и безопасности биоаналога инфликсимаба BCD055 и референтного инфликсимаба у пациентов с анкилозирующим спондилитом: результаты международного многоцентрового рандомизированного двойного слепого клинического исследования III фазы ASART-2. Научно-практическая ревматология 2018;56(3):293–301 [Lila AM, Mazurov VI, Zonova EV, et al. Comparative evaluation of the long-term efficacy and safety of the infliximab biosimilar BCD-055 and reference infliximab in patients with ankylosing spondylitis: results of the international multicenter randomized doubleblind Phase III clinical study ASART-2. Nauchno-Prakticheskaya Revmatologiya = Rheumatology Science and Practice 2018;56(3):293-301 (In Russ.)].
- Lila AM, Mazurov VI, Denisov LN, et al. A phase III study of BCD‐055 compared with innovator infliximab in patients with active rheumatoid arthritis: 54‐week results from the LIRA study. Rheumatol Int 2019;39:1537.
- Коротаева Т.В., Самцов А.В., Бакулев А.Л. и др. Сравнительная эффективность и безопасность биоаналога адалимумаба (BCD-057) и оригинального адалимумаба у пациентов с вульгарным псориазом. Результаты BCD-0572/CALYPSO – международного многоцентрового рандомизированного двойного слепого клинического исследования III фазы. Современная ревматология 2018;12(4):71–84 [Korotaeva TV, Samtsov AV, Bakulev AL, et al. Comparative efficacy and safety of adalimumab biosimilar (BCD-057) and innovator in patients with psoriasis vulgaris. Results of the BCD-057-2/CALYPSO phase III international, multicenter, randomized double-blind clinical trial. Sovremennaya Revmatologiya = Modern Rheumatology Journal 2018;12(4): 71–84 (in Russ.)].