Эффективность семейного скрининга при болезни Фабри в Российской популяции
Изучение эффективности семейного скрининга и препятствий к его проведению у пациентов с болезнью Фабри (БФ).
В ретроспективное исследование включали всех пациентов с диагнозом БФ, подтвержденным при молекулярногенетическом исследовании. На основании опроса определяли индексного пациента (пробанда) и его родственников, которые могли унаследовать мутантный ген. Анализировали родословные для каждого пробанда.
Были обследованы 66 пробандов с БФ, включая 58 мужчин и 8 женщин. У 31 (46,9%) пациента диагноз был установлен во время скрининга, который проводился в российских диализных отделениях. Только один пробанд был ребенком 6 лет, в то время как все остальные пациенты были в возрасте старше 18 лет. В целом у пробандов имелось 488 живых родственников (в 1-4 поколениях), которые могли быть носителями мутантного гена. БФ была диагностирована у 141 (48,3%) из 292 обследованных членов семей пробандов, включая 28 детей. Среднее число пациентов с БФ на одного пробанда составило 2,2±0,3. Основным препятствием к тестированию было отсутствие контактов между членами семьи.
Тестирование родственников индексных пациентов с БФ, диагностированной на основании симптомов или выяв ленной в результате скрининга в группах риска, позволяет идентифицировать еще в среднем 2 пациентов с БФ на одного пробанда и рекомендовать патогенетическое лечение рекомбинантными препаратами a-галактозидазы А на более раннем этапе, в том числе в детском и подростковом возрасте.
Болезнь Фабри (БФ) – это редкая Хсцепленная лизосомная болезнь накопления, которая развивается в результате дефицита фермента α-галактозидазы А, разрушающей гликосфинголипиды в лизосомах. Причиной снижения или отсутствия активности фермента являются мутации гена GLA, расположенного на Х-хромосоме. Накопление гликосфинголипидов, прежде всего глоботриаозилцерамида (GL3) и глоботриаозилсфингозина (LysoGL3), в различных тканях, приводит к прогрессирующему повреждению жизненноважных органов, в том числе почек, сердца и головного мозга, и необратимому ухудшению их функции [1].
Первые симптомы БФ появляются в детском или подростковом возрасте и включают в себя нейропатическую боль, ангиокератомы, снижение или полное отсутствие потоотделения и желудочно-кишечные нарушения (боли в животе, тошнота, рвота, поносы, запоры). Еще одно характерное раннее проявление болезни Фабри – вихревидная кератопатия (cornea verticillata) – коричнево-золотистые отложения в роговице в виде изогнутых линий, которые не приводят к нарушениям зрения, но имеют важное диагностическое значение [2]. Позднее (в возрасте 20-40 лет) у пациентов с БФ появляются признаки поражения внутренних органов, в том числе почек (протеинурия, снижение расчетной скорости клубочковой фильтрации – рСКФ), сердца (гипертрофия миокарда) и головного мозга (инсульт и транзиторные ишемические атаки) [3]. У гемизиготных мужчин клинические проявления БФ развиваются раньше и более выражены, чем у гетерозиготных женщин, хотя у последних также могут наблюдаться как классические симптомы заболевания, так и тяжелое поражение внутренних органов [3].
Своевременный диагноз БФ имеет важное значение, так как ферментозаместительная терапия (ФЗТ) рекомбинантными препаратами a-галактозидазы А (агалсидазой альфа или агалсидазой бета) позволяет уменьшить нейропатическую боль и предупредить или по крайней мере задержать прогрессирующее повреждение органов [4]. Однако диагноз БФ часто устанавливают поздно, иногда спустя несколько десятилетий после появления первых симптомов заболевания, что отражает низкую информированность врачей о редких заболеваниях и/или отсутствие ранних классических симптомов, в частности у женщин или больных с поздним (атипичным) фенотипом, который характеризуется поражением одного органа, прежде всего сердца.
Пациенты с недиагностированной БФ могут быть выявлены путем скрининга в группах риска, например, среди больных с терминальной хронической почечной недостаточностью, получающих лечение диализом, гипертрофией левого желудочка неясного проихождения или инсультом, развившимся в молодом возрасте (до 60 лет). Подобные программы высоко затратны, так как частота диагностики БФ в указанных группах обычно не превышает 1% [5], т.е. чтобы диагностировать БФ у 1 больного, необходимо обследовать по крайней мере 100-200 пациентов или более. Тем не менее, скрининг в группах высокого риска открывает возможность семейного скрининга, позволяющего установить диагноз у родственников больных, в том числе детей, и своевременно начать ФЗТ. Обследование родственников повышает также эффективность затрат на скрининг в группах риска.
Целью исследования было изучение эффективности семейного скрининга и потенциальных препятствий к его проведению у пациентов с БФ.
Материал и методы
В ретроспективное исследование включали всех пациентов с подтвержденным диагнозом БФ, обследованных в клинике им. Е.М. Тареева. Все пациенты дали информированное согласие на участие в исследовании, протокол которого был одобрен локальным этическим комитетом.
Критерии диагноза БФ включали в себя снижение или отсутствие активности a-галактозидазы и мутацию гена GLA в сочетании по крайней мере с одним классическим симптомом (нейропатическая боль, ангиокератомы, cornea verticillata) и/или повышением содержания Lyso-GL3 и/или наличием родственника с определенным диагнозом БФ [6]. Нейропатическая боль (ангиопарестезии) при БФ обычно появляется в детском или подростковом возрасте и характеризуется эпизодами жгучей боли в кистях и стопах, которые возникают в жаркую погоду, при физической нагрузке, повышении температуры тела, быстрой смене температуры окружающей среды. Иногда отмечаются практически постоянные боли, которые, как правило, усиливаются под действием указанных факторов. В жаркую погоду пациенты стараются находиться в тени, а также опускают кисти и/или стопы в холодную воду, чтобы уменьшить боль. С возрастом нейропатическая боль иногда уменьшается или полностью проходит. Ангиокератомы представляют собой скопления мелких темно-красных мягких узелков на передней брюшной стенке, в частности внутри или вокруг пупка, в паховой области, на ягодицах, верхних конечностях, губах. Высыпания чаще распространенные, однако иногда они могут быть единичными, например, внутри пупка, на слизистой оболочке полости рта или половых губах. Наличие вихревидной кератопатии оценивал офтальмолог путем осмотра с помощью щелевой лампы.
Активность a-галактозидазы А измеряли в высушенных пятнах крови с помощью валидированной жидкостной хроматографии – тандемной масс-спектрометрии, а содержание Lyso-GL3 – с помощью тандемной масс-спектрометрии. Исследования проводились в лабораториях Centogene AG (Росток, Германия), ARCHIMED Life Science GmbH (Вена, Австрия), Медико-генетического научного центра имени академика Н.П. Бочкова и/или Национального медицинского исследовательского Центра Здоровья Детей.
Всех больных опрашивали, чтобы установить индексного пациента (пробанда) и его родственников, которые могли унаследовать мутантный ген с учетом Х-сцепленного типа наследования. При необходимости связывались с родственниками больного, чтобы получить более подробную информацию о семье. На основании собранной информации строили и анализировали родословные для каждого пробанда. Оценивали следующие показатели: общее число родственников, которые могли унаследовать мутантный ген, число протестированных на БФ родственников и число выявленных пациентов с БФ (включая женщин, у которых могли отсутствовать проявления болезни). Умерших родственников включали в анализ только в том случае, если диагноз БФ был подтвержден до смерти. Основным критерием диагноза БФ у родственников пробанда с классическим вариантом заболевания служило наличие той же мутации гена GLA. Учитывали также активность a-галактозидазы А (у мужчин), уровень Lyso-GL3 и наличие клинических проявлений.
Статистический анализ был описательным. Нормаль ность распределения проверяли с помощью метода Колмогорова–Смирнова. Для количественных переменных рассчитывали медиану и интерквартильный размах, для качественных – частоту. Количественные переменные сравнивали с помощью U-критерия Манна–Уитни, качественные – с помощью точного метода Фишера. Анализ проводили с помощью программы IBM SPSS Statistics 22.
Результаты
Характеристика индексных пациентов. Нами были идентифицированы 66 пробандов с определенным диагнозом БФ, включая 58 мужчин и 8 женщин (табл. 1). У 31 (46,9%) пациента диагноз был установлен во время скрининга, который проводился в российских диализных отделениях. Только один пробанд был ребенком 6 лет, в то время как все остальные пациенты были в возрасте старше 18 лет.
| Показатели | Мужчины (n=58) | Женщины (n=8) | p |
|---|---|---|---|
| Возраст, лет | 37 [31;50] | 32 [31;50] | 0,86 |
| Возраст на момент появления симптомов, лет | 10 [6;15] | 11 [8;17] | 0,69 |
| Возраст на момент установления диагноза, лет | 33 [29;48] | 30 [26;38] | 0,52 |
| Срок до установления диагноза, лет | 21 [11;29] | 20 [11;17] | 0,94 |
| Ранние проявления, n (%) | |||
| Нейропатическая боль | 42 (73,7) | 6 (75,0) | 0,62 |
| Ангиокератомы | 30 (51,7) | 2 (25,0) | 0,15 |
| Ангидроз/гипогидроз | 38 (65,5) | 2 (25,0) | 0,04 |
| Желудочно-кишечные нарушения | 13 (22,4) | 0 | 0,15 |
| Поражение почек, n (%) | |||
| Альбуминурия/протеинурия | 51 (87,9) | 8 (100) | 0,39 |
| рСКФ<60 мл/мин/1,73 м2 | 3 (5,2) | 2 (25,0) | 0,11 |
| Терминальная ХПН | 31 (53,4) | 0 | 0,004 |
| Диализ | 31 (53,4) | 0 | 0,004 |
| Трансплантация почек | 3 (5,2) | 0 | 0,67 |
| Поражение сердца, n (%) | |||
| Гипертрофия левого желудочка (эхокардиография и/или МРТ) | 43 (74,2) | 2 (25,0) | 0,01 |
| Клинически значимые аритмии | 6 (10,3) | 0 | 0,45 |
| Цереброваскулярная болезнь, n (%) | |||
| Очаги на МРТ | 27/48 (56,3) | 2 (25,0) | 0,10 |
| Инсульт | 14 (24,1) | 0 (11,1) | 0,13 |
| Поражение органа зрения, n (%) | |||
| Вихревидная кератопатия | 28/40 (70,0) | 6 (75,0) | 0,57 |
| Катаракта | 6/40 (15,0) | 1 (12,5) | 0,67 |
| Мутации, n (%) | |||
| Миссенс | 33 (56,9) | 3 (37,5) | 0,26 |
| Нонсенс | 9 (15,5) | 4 (50,0) | 0,04 |
| Другие | 16 (27,6) | 1 (12,5) | 0,33 |
| Снижение активности α-галактозидазы А, n (%) | 58 (100) | 3/7 (42,8) | 0,08 |
| Lyso-GL3, нг/мл | 67,7 | 7,2 | 0,005 |
| Смерть, n (%) | 8 (13,8) | 0 | 0,33 |
Миссенс, нонсенс и другие мутации (делеции, сплайсинговые, инсерционные) гена GLA были выявлены у 36 (54,5%), 13 (19,7%) и 17 (25,8%) пробандов, соответственно. Активность a-галактозидазы А была снижена у 58 (100%) мужчин и 3 (42,9%) из 7 женщин, в то время как содержание Lyso-GL3 в высушенных пятнах крови было повышено у всех пациентов. У мужчин оно было значительно выше, чем у женщин.
У большинства пробандов имелись типичные симптомы БФ с детского или подросткового возраста, однако диагноз был установлен через 20,5 лет (медиана) после их появления. К моменту установления диагноза у всех пробандов, исключая ребенка, имелись поражение почек, выраженная гипертрофия левого желудочка и/или инсульт в анамнезе. После установления диагноза у 30 (45,5%) пробандов была начата ФЗТ.
Анализ родословных. В целом у пробандов имелось 488 живых родственников (в 1-4 поколениях), которые могли быть носителями мутантного гена (табл. 2). Число их варьировалось от 1 до 20 на семью (медиана – 7). Около 60% родственников были протестированы на БФ (активность a-галактозидазы А у мужчин, Lyso-GL3 и молекулярно-генетическое исследование у мужчин и женщин). Основным препятствием к тестированию было отсутствие контактов между членами семьи, например, в связи с проживанием в разных городах. Только 28 родственников отказались от генетических тестов по личным причинам. У небольшой части родственников недавно выявленных пациентов результаты тестов не были получены к моменту проведения данного анализа.
| Параметры | Значения |
|---|---|
| Общее число членов семей | 488 |
| Среднее число родственников в семье | 7,6±0,6 |
| Обследованы на БФ, n (%) | 292 (59,8) |
| Не обследованы, n (%) | 196 (40,2) |
| Отсутствие контакта между родственниками | 155 |
| Отказались от генетического исследования | 28 |
| Результаты тестов ожидаются | 13 |
| Диагностирована БФ, n (%) | 141 (48,3) |
| Мужчины | 45 |
| Женщины | 96 |
| Дети <18 лет | 85 |
| Наличие симптомов | 85 |
| Среднее число родственников с впервые установленным диагнозом БФ на пробанда | 2,2±0,3 |
БФ была диагностирована у 141 (48,3%) из 292 обследованных родственников пробандов, в том числе 45 мужчин и 96 женщин. Двадцать восемь больных с вновь установленным диагнозом были детьми или подростками (менее 18 лет). У 85 пациентов симптомы заболевания отсутствовали. Среднее число пациентов с БФ на одного пробанда составило 2,2±0,3. У 31 (21,9%) пациента была начата ФЗТ.
Примером семейного скрининга может служить родословная, приведенная на рис. 1. У пробанда (стрелка) c 7 лет отмечались классические проявления БФ (нейропатическая боль, гипогидроз, ангиокератомы на стопах). В 26 лет выявлены протеинурия до 1 г/л и микрогематурия, однако к нефрологу не обращался. В 30 лет в связи с нарастанием протеинурии до 2,6 г/сут и снижением азотовыделительной функции почек проведена нефробиопсия. По данным морфологического
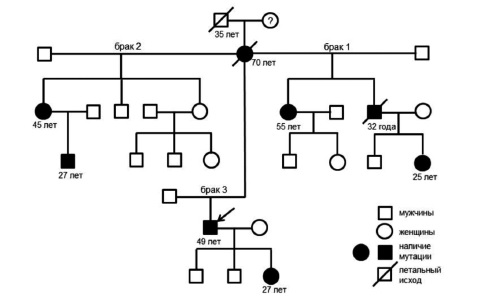
исследования высказано предположение о наличии БФ, однако в 1997 г. возможность проведения молекулярногенетического исследования отсутствовала. В 35 лет у пациента диагностирована терминальная хроническая почечная недостаточность, в связи с чем начато лечение программным гемодиализом. Диагноз БФ установлен в возрасте 44 лет при скрининге, проводившемся в российских отделениях гемодиализа. С 45 лет получает ФЗТ. При обследовании у пациента выявлены катаракта Фабри, гипертрофия миокарда с участками фиброза и очаговые изменения белого вещества головного мозга.
Проведен семейный скрининг: дед умер в 35 лет от заболевания сердца (со слов пациента), брат – в возрасте 32 лет от осложнений терминальной хронической почечной недостаточности. У 27-летней дочери и 25 летней племянницы выявлена такая же мутация гена GLA и снижение активности α-галактозидазы А при отсутствии клинических проявлений БФ. У младшей сестры (45 лет) также обнаружена мутация гена GLA. При сборе анамнеза установлено, что у пациентки с детства отмечались нейропатические боли в кистях и стопах и сниженное потоотделение. Пациентке рекомендовано начало ФЗТ. Старшая сестра (55 лет) также с 10 лет отмечала нейропатические боли в руках и ногах и боли в крупных суставах. При обследовании выявлена та же мутация гена GLA, начальные признаки поражения почек и сердца, начата ФЗТ. Племянник с такой же мутацией гена GLA и сниженной активностью a-галактозидазы А с 5 лет отмечал жгучие боли в кистях и стопах и плохую переносимость жаркой погоды. Пациент получает ФЗТ.
Таким образом, достоверный диагноз БФ был установлен у 5 родственников пробанда, у которого снижение активности a-галактозидазы и мутация гена GLA были выявлены в возрасте 45 лет при скрининге, про водившемся в отделении гемодиализа. Наличие нефропатии Фабри может быть заподозрено при микроскопическом исследовании почечного биоптата, хотя для подтверждения диагноза требуется электронная микроскопия.
Обсуждение
Результаты нашего исследования показали, что семейный скрининг позволяет диагностировать БФ почти у половины родственников, которые потенциально могут быть носителями мутантного гена, или, в среднем, у двух членов семьи индексного больного. Последний показатель был ниже, чем в предыдущем исследовании, проводившемся в 4 американских центрах, занимающихся лизосомными болезнями накопления [7]. Laney и соавт. проводили семейный скрининг в 74 семьях пробандов с БФ. В среднем в каждой семье выявили 5 родственников, страдающих этим заболеванием.
Указанные различия могут объясняться тем, что в российских семьях количество потенциальных кандидатов на тестирование было относительно небольшим (медиана 7). Тем не менее, эффективность семейного скрининга оказалась высокой (48,3%) и значительно превышала таковую скрининга в группах риска. D. Doheny и соавт. провели мета-анализ 67 скрининговых исследований, целью которых было выявление больных с БФ среди пациентов диализных отделений (n=51363), реципиентов почечного трансплантата (n=3074), больных с гипертрофией левого желудочка неясного генеза (n=5491) или инсультом, развившимся в молодом возрасте (n=5978) [5]. Частота патогенных мутаций гена GLA, вызывающих БФ, была сопоставимой в диализных отделениях (0,21% мужчин и 0,15% женщин) и среди больных с инсультом в анамнезе (0,13% мужчин и 0,14% женщин) и несколько выше среди больных c гипертрофией левого желудочка (0,94% мужчин и 0,90% женщин). Сходные данные были получены и в российской программе, в рамках которой у 4289 пациентов, находящихся на лечении программным гемодиализом, определяли активность a-галактозидазы А в высушенных пятнах крови методом тандемной масс-спектрометрии. В случае снижения активности фермента диагноз БФ подтверждали с помощью молекулярно-генетического исследования [8]. Частота БФ составила 0,41%, в том числе 0,64% среди мужчин и 0,06% среди женщин. Среди мужчин самой высокой распространенность БФ была в возрастной группе 40-49 лет и самой низкой в возрастной группе 50-59 лет.
Несмотря на низкую эффективность и, соответственно, высокую стоимость программ скрининга в группах риска, у многих пациентов только они позволяют установить диагноз БФ. Например, в российской популяции примерно у половины индексных пациентов БФ была диагностирована в рамках скрининга, проводившегося в диализных отделениях (следует отметить, что все необходимые исследования были бесплатными для пациента и лечебного учреждения), хотя у большинства из них на протяжении многих лет отмечались симптомы, такие как нейропатическая боль и ангиокератомы, характерные для этого заболевания. При этом медиана срока от появления первых симптомов до установления диагноза составила около 20 лет. Сходные данные приводят и зарубежные исследователи. Например, по данным регистра Fabry Outcome Survey (FOS), у взрослых пациентов с БФ, диагностированной в 2007-2013 гг., медиана срока от появления симптомов до установления диагноза составила 10,5 лет [9]. Она снизилась по сравнению с предыдущим 5-летним периодом (медиана 14,0 лет), однако разница между двумя показателями не достигла статистической значимости.
Хотя у большинства мужчин с классическим вариантом БФ наблюдаются типичные ранние симптомы БФ, они часто интерпретируются неправильно. Например, некоторые проявления БФ, такие как кожная сыпь, нейропатия, повышение температуры тела, изменения со стороны почек, системность поражения внутренних органов, могут имитировать ревматические заболевания. По данным клиники им. Е.М. Тареева, по крайней мере у каждого четвертого пациента с БФ на том или ином этапе развития заболевания устанавливали различные ревматологические диагнозы, в том числе системные васкулиты, ревматоидный (юношеский) артрит, системную красную волчанку, ревматическую лихорадку, болезнь Ослера-Вебера-Рандю, периодическую болезнь [10]. Дополнительные проблемы при проведении дифференциального диагноза создает тот факт, что при БФ могут наблюдаться не только акропарестезии, но и боли в суставах и лихорадка, сопровождающиеся лабораторными признаками воспаления, в частности стойким повышением СОЭ и уровня С-реактивного белка. Например, среди обследованных нами пациентов частота болей в суставах составила 7%, а лихорадки – 16%. Некоторые эксперты даже рекомендуют включать БФ в алгоритм обследования пациентов с лихорадкой неясного генеза [11]. Важное значение для установления правильного диагноза у таких пациентов имеют тщательный анализ клинических проявлений, которые обычно появляются в детском или подростковом возрасте, а также семейного анамнеза, что позволяет заподозрить наличие наследственного заболевания.
Помимо высокой эффективности, достоинством семейного скрининга является возможность своевременного установления диагноза на раннем этапе, когда еще нет необратимого поражения внутренних органов. Например, в нашем исследовании почти 20% пациентов с БФ, выявленной при семейном скрининге, были детьми или подростками, а почти у 40% пациентов отсутствовали симптомы заболевания. В ряде клинических исследований показана более высокая эффективность ранней ФЗТ по сравнению с более поздней. В рандомизированном клиническом исследовании было сопоставлено время до первого клинического исхода (почечного, кардиального, цереброваскулярного или смерти) у 82 больных с БФ с поражением почек, получавших агалсидазу бета в дозе 1 мг/кг или плацебо в течение до 35 мес (медиана 18,5 мес) [12]. Лечение агалсидазой бета задерживало развитие неблагоприятных исходов по сравнению с плацебо, причем эффект лечения у пациентов с расчетной скоростью клубочковой фильтрации >55 мл/мин/1,73 м2 был более выраженным, чем у пациентов с низкими значениями этого показателя. В другом исследовании у 213 пациентов с БФ, получавших агалсидазу бета в дозе 1 мг/кг каждые 2 недели в течение по крайней мере 2 лет, увеличение срока от появления симптомов до начала ФЗТ ассоциировалось с достоверным повышением риска прогрессирования поражения почек (отношение шансов 19; 95% ДИ 2-184; р = 0,0098) [13]. Сходные данные были получены и при анализе динамики массы миокарда левого желудочка в течение 2 лет у 115 мужчин с БФ, получавших агалсидазу бета в дозе 1 мг/кг каждые 2 недели, и 48 нелеченных пациентов с БФ [14]. У мужчин, начавших лечение в возрасте от 18 до <30 лет масса миокарда левого желудочка снижалась в среднем на 3,6 г в год, в то время как при отсутствии лечения у мужчин того же возраста она ежегодно увеличивалась на 9,5 г (p<0,0001). При этом у мужчин, которым ФЗТ была начата в возрасте ≥40 лет, риск прогрессирования гипертрофии левого желудочка был достоверно выше, чем у мужчин, начавших лечение в более молодом возрасте (отношение шансов 5,03; 95% ДИ 1,03-24,49; р=0,047). Замедление нарастания толщины межжелудочковой перегородки на фоне более ранней ФЗТ агалсидазой бета выявили также D. Germain и соавт. в 10-летнем исследовании у 58 пациентов с БФ [15].
При планировании семейного скрининга необходимо учитывать Х-сцепленный тип наследования БФ, чтобы избежать ненужных исследований. У пробандамужчины необходимо проводить обследование родственников по материнской линии, в то время как женщине мутантный ген может быть передан как от матери, так и отца. У мужчины с БФ сыновья всегда здоровы, в то время как всем дочерям передается Ххромосома с мутантным геном (отсутствие такового указывает на ложное отцовство). У женщины с патогенной мутацией гена GLA вероятность рождения ребенка с БФ, как сына, так и дочери, составляет 50%. Тестировать целесообразно всех потенциальных носителей мутантного гена независимо от наличия и характера симптомов, так как фенотипические проявления у членов одной семьи могут существенно отличаться. У женщин ранние симптомы БФ встречаются реже, чем у мужчин, хотя их отсутствие не исключает возможность поражения почек или сердца или развития инсульта.
Основные препятствия к семейному скринингу включают в себя незаинтересованность врачей, финансовые затраты, культурологические проблемы, отсутствие контактов между родственниками, стигмы, ассоциирующиеся с диагнозом генетического заболевания, отсутствие необходимой инфраструктуры и т.д. Тем не менее, в российской популяции были протестированы около 2/3 родственников пробандов. Чаще всего скрининг не удавалось провести из-за отсутствия связей между членами семьи, проживающими в разных городах. Только 14% потенциальных носителей мутантного гена отказались от тестирования по различным причинам. Следует отметить, что в России стоимость тестов не является препятствием к семейному скринингу, так как они являются бесплатными для врачей и медицинских учреждений, в то время как проблемы с инфраструктурой компенсируются эффективной логистикой, позволяющей доставить высушенные пятна крови в центральную лабораторию из любого города.
Результаты нашего исследования свидетельствуют о том, что информированность врачей о БФ остается низкой. В настоящее время в России идентифицировано около 230 пациентов с БФ, т.е. расчетная распространенность заболевания в общей популяции составляет примерно 1 на 600000, в то время как истинный показатель должен быть по крайней мере в 10 раз выше. Более чем у 70% индексных пациентов имелись ранние симптомы БФ, такие как нейропатическая боль, ангиокератомы и/или снижение потоотделения, с детского или подросткового возраста. Однако диагноз БФ никогда не обсуждался даже у пациентов с типичными клиническими симптомами, имеющих родственников со сходными проявлениями. Важное значение имеет распространение информации о болезни Фабри среди педиатров, учитывая тот факт, что только у одного пробанда диагноз был установлен в детском возрасте.
Заключение
Тестирование родственников индексных пациентов с БФ, диагностированной на основании симптомов или выявленной в результате скрининга в группах риска, позволяет идентифицировать еще в среднем 2 пациентов с БФ на одного пробанда и рекомендовать патогенетическое лечение рекомбинантными препаратами a-галактозидазы А на более раннем этапе, в том числе в детском и подростковом возрасте, когда отсутствует необратимое поражение органов-мишеней. В идеале каждого пациентов с вновь диагностированной БФ следует направлять на консультацию к генетику для подробного анализа родословной. Однако эту задачу может выполнить любой врач, который готов потратить свое время на опрос индексного пациента и его родственников.
Используемые источники
- Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 2017;91(2):284293.
- Моисеев С.В., Исмаилова Д.С., Моисеев А.С. и др. Вихревидная кератопатия (cornea verticillata) при болезни Фабри. Терапевтический архив 2018;12:17-22 [Moiseev S, Ismailova D, Moiseev A, et al. Cornea verticillata in Fabry disease. Terapevticheskij arhiv 2018;12:17-22 (In Russ.)].
- Germain DP. Fabry disease. Orphanet J Rare Dis 2010;5:30.
- Germain DP, Charrow J, Desnick RJ, et al., Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet 2015;52:353–8.
- Doheny D, Srinivasan R, Pagant S, et al. Fabry Disease: prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995-2017. J Med Genet 2018;55(4):261-8.
- Smid BE, van der Tol L, Cecchi F, et al. Uncertain diagnosis of Fabry disease: Consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int J Cardiol. 2014;177(2):400-8.
- Laney DA, Fernhoff PM. Diagnosis of Fabry disease via analysis of family history. J Genet Couns 2008;17(1):79-83.
- Моисеев С.В., Намазова-Баранова Л.С., Савостьянов К.В. и др. Распро страненность и клинические проявления болезни Фабри у диализных пациентов. Клин фармакол тер 2017;26(2):27-33 [Moiseev S, Namazova-Baranova LS, Savostyanov KV, et al. Prevalence and clinical features of Fabry disease in dialysis patients. Clin Pharmacol Ther = Klinicheskaya farmakologiya i terapiya 2017;26(2):27-33 (In Russ.)].
- Reisin R, Perrin A, García-Pavía P. Time delays in the diagnosis and treatment of Fabry disease. Int J Clin Pract 2017;71(1). doi: 10.1111/ijcp.12914.
- Моисеев С.В., Новиков П.И., Буланов Н.М. и др. Болезнь Фабри в практике ревматолога. Клин фармакол тер 2018;27(1):39-46 [Moiseev S, Novikov P, Bulanov N, et al. Fabry disease in rheumatology practice. Clin Pharmacol Ther = Klinicheskaya farmakologiya i terapiya 2018;27(1):39-46 (In Russ.)].
- Manna R, Cauda R, Feriozzi S, et al. Recommendations for the inclusion of Fabry disease as a rare febrile condition in existing algorithms for fever of unknown origin. Intern Emerg Med 2017;12(7):1059-67.
- Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146(2):77-86.
- Warnock DG, Ortiz A, Mauer M, et al. Renal outcomes of agalsidase beta treatment for Fabry disease: role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant 2012;27(3):1042-9.
- Germain DP, Weidemann F, Abiose A, et al. Analysis of left ventricular mass in untreated men and in men treated with agalsidase-b: data from the Fabry Registry. Genet Med. 2013;15(12):958-65.
- Germain DP, Charrow J, Desnick RJ, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet 2015;52:353–8.