Цистиноз: патогенез, клинические проявления и лечение
Цистиноз – это редкое наследственное заболевание, передающееся по аутосомно-рецессивному типу и характеризующееся накоплением цистина в лизосомах клеток различных органов и тканей. Нефропатический цистиноз проявляется синдромом Фанкони, развивающимся в раннем детском возрасте, и в конечном итоге приводит к развитию терминальной хронической почечной недостаточности. В подростковом и старшем возрасте у пациентов с цистинозом развиваются различные внепочечные проявления, в том числе поражение поджелудочной, щитовидной и половых желез, увеличение печени и селезенки и др. Методом выбора почечной заместительной терапии у детей и подростков с цистинозом является трансплантация почки. Сразу после установления диагноза цистиноза всем больным необходимо назначать терапию цистеамином, который уменьшает отложения цистина в лизосомах, и задерживает развитие или прогрессирование хронической почечной недостаточности и внепочечных проявлений цистиноза. В статье на примере двух наблюдений обсуждаются клинические проявления и лечение нефропатического цистиноза.
С.В. Моисеев. Цистиноз – это редкое аутосомно-рецессивное заболевание, которое является одной из основных причин развития синдрома Фанкони у детей. Цистиноз был описан в 1903 г. швейцарским биохимиком Emil Abderhalden, хотя первого ребенка с этим заболеванием наблюдал другой швейцарец Eduard Kaufmann [1]. Пациент умер в возрасте 21 месяца, а на вскрытии в различных органах были обнаружены массивные отложения аминокислоты цистина. Нес колько позднее голландский патологоанатом George Lignac более подробно описал проявления цистиноза [2]. Причиной этого наследственного заболевания являются мутации гена CTNS, кодирующего белок цистинозин, который переносит аминокислоту цистин через мембрану лизосом. Нарушение функции транспортного белка приводит к накоплению и кристаллизации цистина в лизосомах клеток, прежде всего почек, которые поражаются уже на первом году жизни, а также глаз, щитовидной и поджелудочной желез, половых органов, мышц и центральной нервной системы [3]. В 2010 г. в США цистинозом страдали 1,4% детей, получавших лечение диализом, и 2,1% детей, перенесших трансплантацию почки [4]. Цистиноз относится к орфанным заболеваниям. В Австралии и странах Европы, в том числе Франции, Германии, Дании и Швеции, его распространенность среди живых новорожденных варьировалась от 1:192000 до 1:115000, хотя она была в несколько раз выше в некоторых популяциях, например, в Бретани (Франция) и Квебеке (Канада) [4-9].
Выделяют три формы цистиноза: (1) нефропатическая инфантильная, (2) нефропатическая ювенильная (поздняя, подростковая), (3) ненефропатическая форма взрослых (доброкачественный ненефропатический цистиноз, цистиноз с поражением глаз). Инфантильная нефропатическая форма цистиноза характеризуется тяжелым течением и приводит к развитию терминальной хронической почечной недостаточности в детском возрасте. При ювенильной форме тяжелое поражение почек отмечается позднее (в подростковом возрасте), а при форме взрослых вообще отсутствует. Увеличение выживаемости детей с цистинозом в первую очередь стало результатом почечной заместительной терапии (диализ и трансплантация почки), однако сегодня возможна и патогенетическая терапия цистеамином, который обеспечивает выведение цистина из лизосом и позволяет замедлить прогрессирование патологического процесса, особенно если лечение было начато до развития необратимого поражения внутренних органов.
В.В. Мальцева. К настоящему времени описаны около 120 мутаций гена CTNS, расположенного на хромосоме 17p13.2. В странах Европы и Северной Америки чаще всего встречается делеция 57257 нуклеотидов, которая наблюдается более чем у половины пациентов с цистинозом [10,11]. Данная мутация была выявлена у 14 (42,4%) из 33 российских детей с инфантильным цистинозом [12]. Реже встречались миссенс мутации c.518A>G и c.1015 G>A, которые наблюдались, соответственно, у 6 (18,2%) детей из пяти неродственных семей и 5 (15,2%) пациентов из трех семей. Первая мутация отмечалась в основном в чеченской этнической группе, а вторая – в карачаевской. Более чем у половины детей были выявлены гомозиготные мутации гена CTNS, которые характерны для популяций с высоким процентом близкородственных браков. Семь патогенных мутаций гена CTNS не были описаны ранее [13]. Фенотип цистиноза в значительной степени зависит от типа мутаций гена CTNS. Делеции и другие мутации, сопровождающиеся полным отсутствием функционального белка, вызывают развитие инфантильного цистиноза, в то время как ювенильная и взрослые формы заболевания ассоциированы с более "легкими" мутациями, в частности, миссенс [14]. В российской выборке концентрации цистина в лейкоцитах у пациентов с гомозиготными стоп-кодонами (делеция 57 тыс нуклеотидов, нонсенс мутации c.283G>T, c.433C>T и c.785G>A) были значительно выше, чем у детей с гомозиготными миссенс мутациями (c.518A>G, c.627C>A и c.1015G>A) [12].
С.В. Моисеев. Каковы клинические проявления цистиноза? Когда следует подозревать это заболевание у ребенка/подростка или взрослого?
Н.В. Чеботарева. В зависимости от возраста, когда появляются симптомы цистиноза, и тяжести поражения почек выделяют три клинических варианта заболевания [3,15]. Чаще всего (95% больных) встречается инфантильный нефропатический цистиноз, который характеризуется развитием синдрома Фанкони уже на первом году жизни. Этот синдром, описанный в начале 30-х гг. Fanconi в Швейцарии, de Toni в Италии и Debré во Франции, встречается как у детей, так и взрослых и развивается в результате дисфункции проксимальных почечных канальцев, которая приводит к увеличению экскреции с мочой аминокислот, глюкозы, фосфора, бикарбоната, натрия, калия, кальция, мочевой кислоты и карнитина. Все перечисленные вещества в норме реабсорбируются в этом сегменте нефрона. Реже выявляется дисфункция дистальных канальцев – гипокалиемия в сочетании с гипохлоремическим метаболическим алкалозом и повышенной активностью ренина в плазме, которые могут имитировать синдром Бартера. У детей основными причинами синдрома Фанкона являются наследственные метаболические заболевания, прежде всего цистиноз [16]. Аминоацидурия и глюкозурия не сопровождаются клиническим симптомами, в частности, снижением массы тела, хотя наличие глюкозы в моче при отсутствии гипергликемии является важным диагностическим признаком. Типичные лабораторные отклонения при синдроме Фанкони включают в себя также гипокалиемию, метаболический ацидоз, гипофосфатемию, гипоурикемию и реже гипонатриемию, а клинические проявления – жажду, полидипсию и полиурию, эпизоды дегидратации, задержку роста, мышечную слабость и рахит. Из-за ограниченного количества мочевых маркеров синдрома Фанкони диагноз цистиноза может быть пропущен в течение первых месяцев жизни. Причинами гипофосфатемического рахита, резистентного к лечению витамином D, у детей и остеомаляции у взрослых с цистинозом являются фосфатурия, потеря витамин D связывающего белка с мочой и нарушение конверсии витамина D на фоне снижения активности α1-гидроксилазы в проксимальных почечных канальцах. Протеинурия обычно минимальная и характеризуется потерей с мочой низкомолекулярных белков. Развернутые проявления синдрома Фанкони при цистинозе обычно наблюдаются уже в возрасте 6-12 мес. Позднее отмечается постепенное снижение скорости клубочковой фильтрации (СКФ), которое приводит к формированию терминальной стадии хронической болезни почек в возрасте 7-10 лет. В международном ретроспективном исследовании, которое проводилось у 205 больных цистинозом в 80-х гг. прошлого столетия, т.е. до разработки патогенетической терапии, медиана возраста пациентов на момент "почечной смерти" (смерти от уремии или инициации почечной заместительной терапии) составила 9,2 года [17]. При этом возраст всех больных с терминальной уремией превышал 5 лет. Таким образом, цистиноз необходимо исключать во всех случаях хронической почечной недостаточности у детей и подростков. Непосредственной причиной канальцевой дисфункции при цистинозе считают отложение цистина в клетках почечных канальцев, хотя патогенез ее может быть более сложным [18]. Так, в опытах на knock-out мышах, не экспрессирующих ген CTNS, изменения клеток проксимальных почечных канальцев определялись до кристаллизации цистина [19,20]. Интересно, что кристаллы цистина главным образом откладываются в интерстициальных клетках почек, редко в подоцитах, но не в тубулярных клетках. Некоторые наблюдения свидетельствуют против кристаллизации цистина как основного фактора прогрессирования тубулярной атрофии, так как формирование деформации по типу "шеи лебедя" не сопровождается отложением кристаллов в проксимальном тубулярном эпителии. Кроме того, кристаллы цистина продолжают накапливаться в интерстиции аллотрансплантатов, при этом синдром Фанкони никогда не развивается. Не выявлено достоверной связи между накоплением цистина в интерстиции и прогрессированием интерстициального фиброза. Напротив, фиброз интерстиция и гломерулосклероз развиваются в участках без отложения кристаллов цистина [21].
Полагают, что избыток цистина, попадающего в цитоплазму тубулярных клеток проксимальных канальцев, влияет на метаболизм этих клеток, влечет за собой снижение митохондриального АТФ, глутатиона и усиление оксидантного стресса. Нарушение внутриклеточного метаболизма тубулярного эпителия вызывает массивный апоптоз тубулярных клеток.
Значительно реже инфантильного нефропатического цистиноза встречаются ювенильная и взрослая формы заболевания (5% случаев) [22]. При ювенильном цистинозе поражение почек менее тяжелое, чем при инфантильном варианте, а существенная задержка роста отсутствует. Функция почек ухудшается медленнее, а терминальная уремия развивается в старшем возрасте (от 12 до 28 лет). Основные проявления ювенильной формы цистиноза – протеинурия и нефротический синдром в сочетании с парциальными канальцевыми нарушениями, например глюкозурией, в то время как полный синдром Фанкони развивается редко. Поражение глаз и развитие терминальной почечной недостаточности наступает позже. При цистинозе взрослых почки и другие внутренние органы не поражаются, а единственным симптомом является фотофобия, связанная с отложением кристаллов цистина в роговице [23].
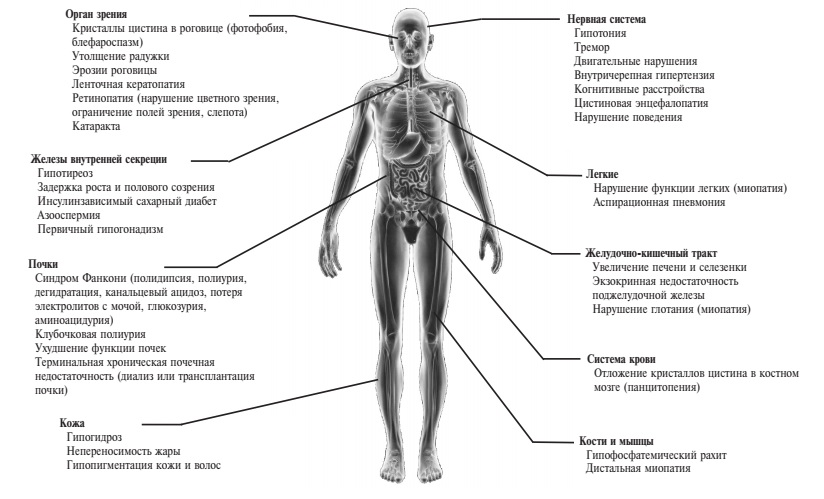
Н.М. Буланов. Цистиноз – это системное заболевание, которое поражает не только почки, но и другие органы и ткани, особенно если пациент не получает патогенетическую терапию [24]. Накопление цистина в лизосомах любых клеток начинается в детском возрасте, однако системные проявления отмечаются позднее нефропатии (рис. 1). Первое внепочечное проявление цистиноза – отложение кристаллов цистина в роговице, которое сопровождается развитием фотофобии и блефароспазма в подростковом или молодом возрасте [25]. К возрасту 16 мес кристаллы цистина определяются в роговице в виде игольчатых опалесцирующих помутнений при осмотре с помощью щелевой лампы у всех пациентов с цистинозом. Цистин может откладываться и в других отделах глазного яблока, в том числе сетчатке, что приводит к слепоте у 10-15% больных [26]. Прогрессирующее накопление и образование кристаллов цистина в фолликулярных клетках щитовидной железы вызывают их атрофию и фиброз и развитие первичного гипотиреоза на втором десятилетии жизни у 50-70% больных цистинозом. Поражение поджелудочной железы при этом заболевании сопровождается нарушением ее экзокринной и эндокринной функции. К возрасту 18 лет у половины пациентов с цистинозом наблюдается постепенное снижение секреции инсулина с развитием сахарного диабета [27]. Примерно у трети больных инфантильным цистинозом отмечается увеличение печени и/или селезенки, хотя функция печени обычно не изменяется. У мальчиков часто развивается первичный гипогонадизм, однако у женщин репродуктивная функция, как правило, сохраняется [28]. Поражение потовых желез приводит к сниженному потоотделению и непереносимости жары. У детей с инфантильным цистинозом, доживших до старшего возраста благодаря почечной заместительной терапии, обычно наблюдаются последствия хронической почечной недостаточности или осложнения длительной иммуносупрессивной терапии после трансплантации почки. Поражение центральной нервной системы является поздним осложнением цистиноза, развивающимся в течение третьего десятилетия жизни. Различают два основных проявления цистиновой энцефалопатии – преобладание мозжечковых и пирамидных нарушений и поражение головного мозга с эпизодами, сходными с острым нарушением мозгового кровообращения. Интеллект у больных цистинозом сохранен, но может наблюдаться изменение нейрокогнитивных функций, таких как зрительная память, внимание, планирование и скорость движений. У детей поражение нервной системы может проявляться синдромом дефицита внимания и трудностями в обучении. Наиболее частыми находками при компьютерной томографии являются атрофия коры головного мозга и кальциноз базальных ганглиев. К поздним жизнеугрожающим осложнениям цистиноза относится вакуольная миопатия, возникающая в результате накопления цистина в мышечных волокнах. Пациенты страдают от прогрессирующей атрофии мышц и снижения мышечной силы. Отмечается атрофия мышц кистей (тенара, гипотенара, межкостных мышц) с характерным положением кисти в виде "когтя". Наблюдаются симптомы оро-моторной дисфункции, такие как дисфагия, нарушение речи, сиплый голос, отсутствие рвотного рефлекса. Особенно опасно поражение дыхательных мышц с развитием рестриктивной дыхательной недостаточности. В табл. 1 приведена частота различных внепочечных проявлений цистиноза у 100 больных в возрасте от 18 до 45 лет, большинство из которых перенесли трансплантацию почки (92%) [29]. В этом исследовании умерли 33 больных в возрасте в среднем 28,5 лет. Причины смерти включали в себя сепсис (у 9), уремию (у 5), пневмонию (у 4), портальную гипертензию (у 3) и др.
| Проявления | n/N (%) |
|---|---|
| Гипотиреоз | 92/100 (92) |
| Гипогонадизм (у мужчин) | 39/53 (75) |
| Нарушение функции легких | 53/77 (69) |
| Нарушение глотания | 58/97 (60) |
| Миопатия | 50/100 (50) |
| Ретинопатия | 32/100 (32) |
| Инсулинзависимый сахарный диабет | 33/100 (33) |
С.В. Моисеев. В нашей стране самый большой опыт обследования и лечения пациентов с цистинозом накоплен в нефрологическом отделении НМИЦ здоровья детей. Сколько таких больных сегодня наблюдается в отделении? Как проявлялось заболевание?
А.Н. Цыгин. За период с 2008 по 2020 год диагноз нефропатического цистиноза был установлен у 34 детей, в том числе 20 мальчиков (58,8%) и 14 девочек (41,2%). Эти пациенты проживают в 21 субъекте и 7 федеральных округах Российской Федерации. Мини мальный возраст на момент постановки диагноза составил 8 мес, максимальный – 13 лет (медиана 2,2 [1,9; 6,4] года). Наследственный анамнез был отягощен в 6 (17,6%) семьях. У всех детей была выявлена задержка физического развития. На первом году жизни частыми клиническими симптомами были полидипсия (38%), полиурия (38%), глюкозурия (38%), гипокалиемия (29%) и гипофосфатемия (14%). К возрасту 2 лет формирование рахитических изменений нижних конечностей отмечено у 14 (41%) детей. При осмотре с помощью щелевой лампы отложения кристаллов цистина в роговице выявлены в 28 (82%) случаев. Возраст достижения ХБП 3 стадии у 22 (32,4%) пациентов соответствовал 5,2±3,0 годам. В среднем возраст достижения ХБП 5 стадии составил 8,0±2,0 лет. 18 детей (52,9%) достигли терминальной стадии почечной недостаточности. Из них у 12 (35,3%) пациентов выполнена трансплантация почки. У всех сохраняется хорошая функция трансплантата. За период наблюдения умерли 3 (8,8%) детей в возрасте от 6 до 10 лет.
С.В. Рощупкина. Естественное течение инфантильного цистиноза иллюстрирует следующее клиническое наблюдение. Пациентка Л., 34 лет, была госпитализирована в клинику им. Е.М. Тареева в 2019 г. С раннего детского возраста отмечалась задержка роста, а при обследовании выявлен синдром Фанкони – фосфатурия, кальцийурия, глюкозурия, проксимальный канальцевый метаболический ацидоз, гипофосфатемический рахит с гипокальциемией. В возрасте 5 лет отмечено нарушение функции почек, а в 9 лет была начата заместительная почечная терапия в связи с развитием терминальной почечной недостаточности. В 14 лет проведена аллогенная трансплантация почки. С этого же времени отмечает светобоязнь и постепенное снижение зрения. В 24 года при осмотре офтальмологом с помощью щелевой лампы обнаружены отложения кристаллов цистина в роговице. На основании характерного поражения почек и роговицы установлен диагноз цистиноза. Рекомендована патогенетическая терапия цистеамином внутрь и в виде глазных капель. Однако лечение пациентка не получала еще в течение 10 лет после диагностики заболевания. В возрасте 32 лет выявлены гипотиреоз и сахарный диабет с гликемией натощак до 15,6 ммоль/л и повышением содержания гликированного гемоглобина до 8,8%. Начата заместительная терапия L-тироксином и инсулином. Кроме того, отмечено увеличение печени и селезенки. На протяжении всего времени наблюдения сохранялась стабильная, умеренно сниженная функция почечного трансплантата (сывороточное содержание креатинина 160 мкмоль/л, скорость клубочковой фильтрации в пробе Реберга – 41 мл/мин).
В возрасте 33 лет пациентка перенесла острый инфаркт миокарда без подъема сегмента ST. При коронарографии выявлено многоуровневое атеросклеротическое поражение коронарных артерий, в связи с чем проведена чрескожная транслюминальная баллонная ангиопластика со стентированием огибающей артерии. При магнитно-резонансной томографии головного мозга обнаружен кальциноз церебральных артерий.
В возрасте 34 лет при обследовании в клинике отмечались резкое отставание в физическом развитии (рост – 132 см, масса тела – 37 кг), тяжелое поражение органа зрения (помутнение роговицы и полная слепота), декомпенсированный сахарный диабет, гепатоспленомегалия и тромбоцитопения. Диагноз: инфантильный цистиноз с поражением почек (синдром Фанкони, аллогенная трансплантация почки), органа зрения (трофические язвы, кальцинаты роговицы, атрофия зрительного нерва, слепота), поджелудочной железы (инсулинзависимый сахарный диабет), щитовидной железы (гипотиреоз), центральной нервной системы (атеросклероз, кальциноз церебральных артерий), инфаркт миокарда передней стенки левого желудочка без подъема сегмента ST, атеросклероз коронарных артерий, баллонная ангиопластика со стентированием огибающей артерии. Начата терапия цистеамином в дозе 50 мг/кг/сут с удовлетворительной переносимостью.
А.Н. Цыгин. В представленном наблюдении у пациентки с раннего детского возраста определялись задержка роста и проявления синдрома Фанкони, в том числе гипофосфатемический рахит, а в возрасте 9 лет была начата почечная заместительная терапия. Цистиноз может быть выявлен по крайней мере у каждого пятого ребенка с синдромом Фанкони и примерно у каждого двадцатого ребенка с терминальной уремией, нуждающегося в почечной заместительной терапии. Однако этот диагноз не обсуждался даже после появления еще одного характерного симптома цистиноза – светобоязни, связанной с накоплением кристаллов цистина в роговице. Цистиноз был диагностирован только в возрасте 24 лет, т.е. спустя более 20 лет после появления первых симптомов заболевания, когда при осмотре с помощью щелевой лампы были выявлены отложения цистина в роговице. Как указано выше, у всех больных инфантильным цистинозом кристаллы цистина определяются в роговице уже к возрасту 1,5 лет. К сожалению, пациентка на протяжении 10 лет после установления диагноза не получала патогенетическую терапию цистеамином в связи с отсутствием препарата в Российской Федерации. На этом фоне отмечалось прогрессирование заболевания с развитием слепоты, инсулинзависимого сахарного диабета, гипотиреоза, увеличения печени и селезенки. Сходные данные приводят и другие авторы. D. Theodoropoulos и соавт. проанализировали течение цистиноза у 36 взрослых пациентов с инфантильным цистинозом (максимальный возраст 36 лет) [30]. Все больные перенесли трансплантацию почки в возрасте от 5 до 19 лет. Только 11 из них получали адекватную терапию цистеамином. Семь (19%) из 36 больных умерли. У большинства пациентов наблюдались тяжелые проявления цистиноза, в том числе слепота (14%), гипотиреоз, требующий заместительной терапии L-тироксином (85%), и инсулинзависимый сахарный диабет (14%). При этом результаты трансплантации почки у больных цистинозом считают очень хорошими. C. Cohen и соавт. сопоставили отдаленные исходы трансплантации почки у 30 взрослых пациентов с цистинозом и 93 больных контрольной группы, подобранных по возрасту и ряду других показателей [31]. В основной группе трансплантация была выполнена в возрасте от 7 до 36,5 лет (медиана – 20,4 года). К этому времени у всех больных имелись отложения цистина в роговице, у 3 – сахарный диабет, у 7 – гипотиреоз. Выживаемость трансплантата у больных цистинозом была выше, чем в контрольной группе (р=0,013), а многофакторный анализ подтвердил, что наличие этого заболевания ассоциируется со снижением риска отторжения трансплантата (отношение рисков 0,11; 95% доверительный интервал 0,020,61). Частота посттрансплантационного сахарного диабета в основной группе была выше, чем в контрольной (13,0% и 5,0%), однако различия не достигли статистической значимости. Высокую выживаемость трансплантата демонстрирует и наше наблюдение 34летней пациентки с цистинозом, перенесшей трансплантацию почки около 20 лет назад. Следует подчеркнуть, что синдром Фанкони в трансплантате никогда не рецидивирует [15]. Соответственно, трансплантацию почки считают методом выбора почечной заместительной терапии у детей с цистинозом. Синдром Фанкони иногда сохраняется после начала диализа или трансплантации почки, что в редких случаях требует удаления нативных почек вследствие чрезмерных потерь жидкости и электролитов [32].
С.В. Моисеев. Как объяснить развитие острого коронарного синдрома у молодой женщины? Поражаются ли коронарные артерии при цистинозе?
Н.В. Чеботарева. Несмотря на молодой возраст, пациентка в течение около 30 лет страдает хронической болезнью почек, которая ассоциируется с увеличением риска сердечно-сосудистых исходов у пациентов не только старшего, но и молодого возраста. В крупном американском исследовании были проанализированы частота госпитализаций по поводу сердечно-сосудистых заболеваний и сердечно-сосудистая смертность более чем у 33000 молодых пациентов с терминальной стадией хронической болезни почек, начавших почечную заместительную терапию (гемодиализ, перитонеальный диализ или трансплантация почки) в возрасте 1-11, 1221 и 21-29 лет [33]. Во всей выборке доля сердечнососудистых причин в структуре общей смертности составила 37,7% и была выше всего в группе пациентов в возрасте 21-29 лет (39,1%). Риск сердечно-сосудистой смерти увеличивался с возрастом и у молодых людей был достоверно выше, чем у подростков и детей. Кроме того, факторами, ассоциировавшимися с повышенным риском смерти от сердечно-сосудистых заболеваний, были женский пол и низкий индекс массы тела, а также различные сопутствующие заболевания, в частности сахарный диабет, в то время как трансплантация почки ассоциировалась с улучшением сердечно-сосудистых исходов по сравнению с лечением диализом. Таким образом, развитие острого коронарного синдрома у молодой женщины с цистинозом не было неожиданным, учитывая большую длительность почечной заместительной терапии, наличие умеренной дисфункции почечного трансплантата и дополнительных факторов риска, таких как низкий индекс массы тела и сахарный диабет.
В.В. Мальцева. У взрослых пациентов с цистинозом нередко определяется кальциноз артерий. В крупном исследовании у 100 больных в возрасте от 18 до 45 лет частота его составила 31%, а кальцинаты в ткани головного мозга определялись в 22% случаев [29]. M. Ueda и соавт. выявили кальциноз сосудов с помощью компьютерной томографии после трансплантации почки у 32% из 41 пациента с цистинозом [34]. У большнства больных кальцинаты локализовались в коронарных артериях. Одному пациенту было выполнено аортокоронарное шунтирование трех артерий в возрасте 25 лет. Паценты с кальцинозом артерий были старше и чаще страдали сахарным диабетом. Частота кальциноза увеличивалась, если пациенты длительно не получали цистеамин. При этом длительность лечения диализом не отличалась между группами больных, у которых определялись и не определялись отложения кальция в стенках артерий. Эти данные могут указывать на роль цистина в развитии кальциноза артерий при цистинозе. Дополнительным фактором риска сердечно-сосудистых исходов у пациентов с цистинозом может быть также гиперхолестеринемия, которая нередко встречается при этом заболевании [35].
С.В. Моисеев. В представленном наблюдении диагноз цистиноза был установлен на основании клинической картины и результатов офтальмологического исследования. Достаточно ли этих данных?
Н.М. Буланов. У нашей пациентки диагноз цистиноза, конечно, не вызывает сомнения, учитывая наличие типичных клинических проявлений, включая тяжелое поражение почек, развившееся в детском возрасте, низкий рост, изменения со стороны органа зрения (в частности светобоязнь), инсулинзависимый сахарный диабет и гипотиреоз. Наличие кристаллов цистина в роговице подтверждает этот диагноз, так как отложения цистина не обнаруживаются при других заболеваниях, сопровождающихся синдромом Фанкони, в том числе тирозинемии, галактоземии, гепаторенальном гликогенозе, болезни Дента, синдроме Лове и т.д. [15]. У детей и подростков осмотр роговицы с помощью щелевой лампы является доступным скрининговым методом диагностики, хотя исследование должно проводиться опытным офтальмологом. Необходимо также учитывать
Е.Ю. Андреева. Приводим еще одно наблюдение цистиноза у 23-летнего мужчины. В возрасте 1 года развился синдром Фанкони, проявлявшийся проксимальным канальцевым гиперхлоремическим метаболическим ацидозом, бикарбонатурией, калийурией, гипокалиемией, кальциурией, фосфатурией. В 7 лет в связи с появлением светобоязни пациент был осмотрен офтальмологом. С помощью щелевой лампы выявлены характерные отложения кристаллов цистина в роговице. Отложения цистина были обнаружены также в интерстиции почки при гистологическом исследовании нефробиоптата. При молекулярно-генетическом исследовании в лаборатории генетики и клеточной биологии НМИЦ здоровья детей выявлена мутация гена CTNS, подтверждавшая диагноз цистиноза. Патогенетическая терапия не проводилась. В 12 лет в связи с развитием терминальной почечной недостаточности выполнена аллогенная трансплантация почки от матери. Непосредственно после трансплантации начата патогенетическая терапия цистеамином (Цистагон 50 мг/кг/сут) внутрь и в виде глазных капель (Цистадропс 1 мл/сут) с хорошей переносимостью. Наблюдался в отделении нефрологии НМИЦ здоровья детей. В 16 лет диагностирован сахарный диабет – уровень гликемии натощак составлял 8,8 ммоль/л (HbA1c – 7,7%). Впервые обследован в клинике им. Е.М. Тареева в возрасте 23 лет. Рост 165 см, масса тела 72 кг. Функция щитовидной железы и острота зрения не снижены. Сохраняется хорошая функция трансплантата (сывороточный уровень креатинина – 124 мкмоль/л, СКФ в пробе Реберга – 75 мл/мин). Пациент продолжает терапию цистеамином. Диагноз: инфантильный цистиноз с поражением почек (синдром Фанкони, аллогенная трансплантация почки от матери), органа зрения (кератопатия, отложение кристаллов в роговице) и поджелудочной железы (сахарный диабет).
О.Н. Науменко. Как и первый случай, представленное наблюдение иллюстрирует типичные ранние проявления инфантильного цистиноза – синдром Фанкони, развившийся в возрасте 1 года, и светобоязнь, появившаяся в возрасте 7 лет. Диагноз был подтвержден результатами осмотра с помощью щелевой лампы, биопсии почки и молекулярно-генетического исследования. Концентра цию цистина в лейкоцитах не измеряли, так как этот метод в нашей стране начали использовать позднее. Функция почечного трансплантата у пациента остается хорошей на протяжении более 10 лет. Через 4 года после трансплантации почки развился сахарный диабет, однако другие внепочечные проявления цистиноза отсутствуют (нормальный рост, не определяются кристаллы цистина в роговице, не снижена функция щитовидной железы), что, несомненно, отражает эффективность длительной терапии цистеамином. Нельзя также исключить вклад иммуносупрессивной терапии глюкокортикостероидами и такролимусом в развитие сахарного диабета.
Н.В. Чеботарева. Ранняя диагностика цистиноза имеет очень важное значение, так как патогенетическая терапия цистеамином улучшает рост у детей и задерживает развитие терминальной хронической болезни почек и большинства внепочечных проявлений заболевания. Цистеамин проникает в лизосомы, расщепляет цистин на две молекулы и соединяется с одной из них с помощью дисульфидного мостика. Цистеин и цистеинциастеаминовый комплекс не нуждаются в цистозине для выхода из лизосом, так как они транспортируются белком PQLC2. Лечение цистеамином, которое следует начинать сразу после установления диагноза цистиноза, не только предупреждает дальнейшее накопление цистина, но и вызывает выведение его из лизосом клеток. Цистеамин производится в виде битартрата для приема внутрь под коммерческим названием Цистагон, а также в виде капель (0,55% раствор цистеамина гидрохлорида). Капли применяют для растворения кристаллов цистина в роговице, так как пероральная терапия цистеамином не оказывает на них влияние. Эффективность местной терапии каплями цистеамина подтверждена при мета-анализе 7 исследований, в том числе двойных слепых, плацебо-контролируемых [37]. H. Liang и соавт. проанализировали отдаленные результаты лечения глазными каплями цистеамина (в среднем 3,3 раза в день) в течение до 45 мес у 130 больных цистинозом [38]. Применение препарата позволяло поддерживать нормальную остроту зрения и вызывало уменьшение количества кристаллов цистина в роговице. Светобоязнь уменьшалась в течение 3 мес. Нежелательные реакции включали в себя преходящее раздражение, покалывание и туман перед глазами. В отдельных случаях развивались кератит и язвы роговицы, однако они могли быть связаны с основным заболеванием.
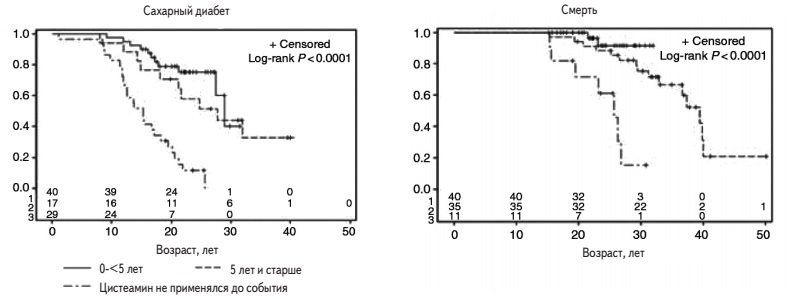
Пероральная терапия цистемином не влияет на течение синдрома Фанкони, однако позволяет задержать развитие терминальной хронической почечной недостаточности на 6-10 лет [3]. Кроме того, она предупреждает или тормозит развитие внепочечных проявлений цистиноза. В когортном исследовании у больных, продолжавших терапию цистеамином более 20 лет, частота сахарного диабета и миопатии снизилась с 28% до 0% и с 60% до 0%, соответственно, а у больных, получавших препарат в течение более 8 лет, частота гипотиреоза снизилась с 87% до 56% [29]. Длительность терапии цистеамином в этом исследовании у 33 умерших пациентов составила 2,1±0,7 года, а у 67 выживших больных – 9,6±0,9 года. Brodin-Sartorius и соавт. изучали эффективность цистеамина в ретроспективном исследовании у 86 взрослых больных (средний возраст 26,7 лет) [39]. 75 из них получали препарат в течение в среднем 17,4 лет. Терапия цистеамином, начатая в возрасте до 5 лет, значительно задерживала развитие терминальной хронической болезни почек, а также гипотиреоза, сахарного диабета и нейромышечных нарушений. У пациентов, начавших лечение после 5 лет, сроки развития сахарного диабета и гипотиреоза, значительно увеличились по сравнению с таковыми у нелеченных пациентов (рис. 2). Кроме того, терапия цистеамином привела к увеличению продолжительности жизни больных цистинозом. Полученные данные свидетельствуют о том, что пероральную терапию цистеамином лучше начинать в раннем детском возрасте, однако она обоснована и в более поздние сроки после установления диагноза, в том числе у взрослых пациентов, у которых лечение по крайней мере позволяет избежать дальнейшего ухудшения состояния. Для контроля эффективности терапии цистеамином целесообразно определять концентрацию цистина в лейкоцитах, которая не должна превышать 1 нмоль 1/2 цистина на мг белка [40].
Е.А. Тао. Все больные цистинозом нуждаются в симптоматической терапии, которая включает в себя восполнение потерь электролитов и жидкости при синдроме Фанкони, применение витамина D при рахите, рекомбинантного гормона роста при задержке роста, тестостерона при гипогонадизме, гормонов щитовидной железы при гипотиреозе, инсулина при сахарном диабете и т.п. [41,42]. Ведение детей с синдромом Фанкони может представлять большие трудности, учитывая значительную потерю электролитов на фоне выраженной полиурии и частой рвоты. В 1982 г. G. Hay cock и соавт. описали 3 детей с цистинозом, у которых индометацин уменьшал полиурию и клинические симптомы [43]. Позднее уменьшение полиурии (на 30-70%) и увеличение прибавки массы тела при лечении индометацином было отмечено и другими авторами [42]. Индометацин ингибирует синтез простагландинов в паренхиме почек, которые увеличивают почечный кровоток, подавляют реабсорбцию хлорида натрия и снижают экспрессию аквапорина 2. Таким образом, индометацин усиливает реабсорцию соли в петле Генле и собирательных канальцах. У здоровых людей препарат не оказывает существенного влияния на СКФ, однако он может вызвать ее снижение у пациентов с нарушенной перфузией почек, так как уве личение продукции простагландинов является компенсаторным механизмом, направленным на поддержание функции почек. Кроме того, индометацин может оказывать токсическое действие на интерстиций почек. В целом индометацин считают относительно безопасным у детей с почечным синдромом Фанкони, однако препарат следует отменить в случае развития дегидратации, артериальной гипотонии или ухудшения функции почек [42]. Ингибиторы АПФ и блокаторы ангиотензиновых рецепторов широко используются для замедления темпа снижения СКФ у пациентов с заболеваниями почек, особенно сопровождающимися протеинурией. Высказано предположение о том, что они могут быть использованы и у детей с нефропатическим цистинозом [44]. Однако эффективность блокаторов ренин-ангиотензиновой системы у таких больных не доказана. Более того, они могут ухудшить перфузию почек, которая обычно снижена у больных с синдромом Фанкони. В связи с этим применять подобные препараты у пациентов с цистинозом следует с осторожностью.
С.В. Моисеев. Важное значение для своевременной диагностики цистиноза, как и любых других редких заболеваний, имеет настороженность врачей, прежде всего педиатров нефрологов. Как показывают наши наблюдения, этот диагноз устанавливают поздно несмотря на наличие типичных проявлений заболевания. Цистиноз следует исключать у всех детей с синдромом Фанкони, а также у больных с терминальной уремией, развивающейся в детском или подростковом возрасте. Подтвердить наличие цистиноза относительно несложно. В качестве скринингового метода диагностики можно использовать осмотр роговицы с помощью щелевой лампы. Высокочувствительными и специфичными диагностическими методами являются измерение концентрации цистина в лейкоцитах и молекулярногенетическое исследование. Всем больным цистинозом показана пожизненная терапия цистеамином для профилактики развития или прогрессирования инвалидизирующих проявлений заболевания.
Используемые источники
- Abderhalden E. Familiare cystindiathese. Z Physiol Chem. 1903;38:557–61
- Lignac GOE. Uber storung des cystinstoffwechsels bei kindern. Deutsch Arch Klin Med 1924;145:139–50.
- Elmonem MA, Veys KR, Soliman NA, et al. Cystinosis: a review. Orphanet J Rare Dis 2016;11:47.
- North American Pediatric Renal Trials and Collaborative Studies. NAPRTCS Annual Reports. NAPRTCS Online [online]. 2011. https://web.emmes.com/ study/ped/annlrept/annlrept.html.
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281:249–54.
- Cochat P, Cordier B, Lacote C, Said M-H. Cystinosis: epidemiology in France. In: Broyer M, editor. Cystinosis. Paris: Elsevier; 1999, 28–35.
- Manz F, Gretz N. Cystinosis in the Federal Republic of Germany. J Inherit Metab Dis 1985;8:2–4.
- Ebbesen F, Mygind KI, Holck F. Infantile nephropathic cystinosis in Denmark. Danish Med Bull 1976;23:216–22.
- Hult M, Darin N, von Döbeln U, Månsson JE. Epidemiology of lysosomal storage diseases in Sweden. Acta Paediatr 2014;103:1258–63.
- Levtchenko E, van den Heuvel L, Emma F, Antignac C. Clinical utility gene card for: cystinosis. Eur J Hum Genet 2014;22(5):e1-3
- Shotelersuk V, Larson D, Anikster Y, et al. CTNS mutations in an Americanbased population of cystinosis patients. Am J Hum Genet 1998;63:1352–62.
- Савостьянов К.В., Мазанова Н.Н., Пушков А.А., и др. Хромато-масс-спектрометрическая и молекулярно-генетическая диагностика цистиноза у российских детей. Педиатрия 2018;97(5):71-8 [Savostyanov KV, Mazanova NN, Pushkov AA, et al. Chromatography-mass spectometry and molecular genetic diagnosis of cystinosis in Russian children. Pediatriya 2018;97(5):71-8 (In Russ.)].
- Savostyanov K, Pushkov A, Zhurkova N, et al. Selective screening for nephropathic cystinosis among high-risk contingents of the children population in Russia. Mol Gen Metab 2020;129(2):S144.
- Attard M, Jean G, Forestier L, et al. Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin. Hum Mol Genet 1999;8:2507–14.
- Цыгин А.Н., Каган М.Ю., Картамышева Н.Н. и др. Нефропатический цистиноз. Недооцененная проблема детской нефрологии. Клиническая нефрология 2011;4:20-3 [Tsygin AN, Kagan Myu, Kartamysheva NN, et al. Nephropathic cystinosis. An underestimated problem in pediatric nephrology. Clinical Nephrology 2011;4:20-3 (In Russ.)].
- Foreman J. Fanconi Syndrome. Pediatr Clin N Am 2019;66:159–167
- Gretz N, Manz F, Augustin R, et al. Survival time in cystinosis. A collaborative study. Proc Eur Dial Transplant Assoc 1983;19:582-9.
- Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol 2017;13(2):115–31.
- Cherqui S, Sevin C, Hamard G, et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol 2002; 22:7622–32.
- Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant 2010;25:1059–66.
- Mahoney СP, Striker GE. Early development of the renal lesions in infantile cystinosis. Pediatr Nephrol 2000;15:50–6.
- Servais A, Morinière V, Grü nfeld JP, et al. Late-onset nephropathic cystinosis: clinical presentation, outcome, and genotyping. Clin J Am Soc Nephrol. 2008;3:27–35.
- Anikster Y, Lucero C, Guo J, et al. Ocular non-nephropathic cystinosis: clinical, biochemical, and molecular correlations. Pediatr Res. 2000;47:17–23.
- Kasimer RN, Langman CB. Adult complications of nephropathic cystinosis: a systematic review. Pediatr Nephrol. 2020 Feb 3. Epub ahead of print.
- Kaiser-Kupfer MI, Caruso RC, Minkler DS, Gahl WA. Long-term ocular manifestations in nephropathic cystinosis. Arch Ophtalmol. 1986;104:706–11.
- Gahl WA, Kuehl EM, Iwata F, et al. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eye drops. Mol Genet Metab 2000;71:100–20.
- Filler G, Amendt P, von Bredow MA, et al. Slowly deteriorating insulin secretion and C-peptide production characterizes diabetes mellitus in infantile cystinosis. Eur J Pediatr 1998;157:738–42.
- Blakey H, Proudfoot-Jones J, Knox E, Lipkin G. Pregnancy in women with cystinosis. Clin Kidney J 2019;12(6):855-8.
- Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med 2007;147(4):242-50.
- Theodoropoulos DS, Krasnewich D, Kaiser-Kupfer MI, Gahl WA. Classic nephropathic cystinosis as an adult disease. JAMA 1993;270(18):2200-4.
- Cohen C, Charbit M, Chadefaux-Vekemans B, et al. Excellent long-term outcome of renal transplantation in cystinosis patients. Orphanet J Rare Dis 2015;10:90.
- Wilmer MJ, Schoeber JP, van den Heuvel L, et al. Cystinosis: practical tools for diagnosis and treatment. Pediatr Nephrol 2011;26:205–15.
- Modi ZJ, Lu Y, Ji N, et al. Risk of cardiovascular disease and mortality in young adults with end-stage renal disease: An analysis of the US Renal Data System. JAMA Cardiol 2019;4(4):353-62.
- Ueda M, O’Brien K, Rosing DR, et al. Coronary artery and other vascular calcifications in patients with cystinosis after kidney transplantation. Clin J Am Soc Nephrol 2006;1:555–62.
- Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol 2008;23:863–78.
- Hohenfellner K, Bergmann C, Fleige T, et al. Molecular based newborn screening in Germany: Follow-up for cystinosis. Mol Genet Metab Rep. 2019;21:100514.
- Kaur S, Sarma P, Kaur H, et al. Efficacy and safety of topical cysteamine in corneal cystinosis: a systematic review and meta-analysis. Am J Ophthalmol 2020;S0002-9394(20)30418-9.
- Liang H, Labbé A, Baudouin C, Plisson C, Giordano V. Long-term follow-up of cystinosis patients treated with 0.55% cysteamine hydrochloride. Br J Ophthalmol 2020;bjophthalmol-2020-316450.
- Brodin-Sartorius A, Tête MJ, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int 2012;81(2):179-89.
- Gahl WA, Schneider JA, Schulman JD, et al. Predicted reciprocal serum creatinine at age 10 years as a measure of renal function in children with nephropathic cystinosis treated with oral cysteamine. Pediatr Nephrol 1990;4:129–35.
- Langman CB, Barshop BA, Deschênes G, et al. Controversies and research agenda in nephropathic cystinosis: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int 2016;89(6): 1192-203. 88
- Emma F, Nesterova G, Langman C, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant. 2014;29 Suppl 4(Suppl 4):iv87-iv94.
- Haycock GB, Al-Dahhan J, Mak RH, Chantler C. Effect of indomethacin on clinical progress and renal function in cystinosis. Arch Dis Child 1982;57:934–9.
- Levtchenko E, Blom H, Wilmer M et al. ACE inhibitor enalapril diminishes albuminuria in patients with cystinosis. Clin Nephrol 2003;60:386–9.