Семейный генетический скрининг при редких наследственных заболеваниях (на примере болезни Фабри)
Семейный генетический скрининг, т.е. обследование родственников пациентов с впервые установленным диагнозом редкого наследственного заболевания, позволяет выявить заболевание на ранней стадии у членов семьи и назначить им специфическую терапию, если таковая доступна. Болезнь Фабри (БФ) – это X-сцепленная лизосомная болезнь накопления, которая может привести к тяжелому поражению почек, сердца и головного мозга. Диагноз БФ обычно устанавливают спустя много лет после появления первых симптомов в связи с низкой информированностью врачей о редких заболеваниях. Одним из подходов к диагностике БФ является скрининг в группах риска среди пациентов, например, с гипертрофией левого желудочка или нефропатией несного происхождения, у которых вероятность наличия этого заболевания значительно выше, чем в общей популяции. На следующем этапе проводится тестирование родственников выявленных пациентов с учетом типа наследования заболевания. Существуют различные барьеры, которые затрудняют семейный скрининг и отличаются в разных странах. К ним относятся финансовые затраты и низкая осведомленность о важности скрининга, географическое разобщение семей, особенности национальной инфраструктуры, нехватка врачей-генетиков и др.
В последние десятилетия постепенно расширяется список редких наследственных заболеваний, при которых возможна эффективная патогенетическая терапия. Примером могут служить некоторые лизосомные болезни накопления, такие как болезни Гоше и Фабри, мукополисахаридозы и болезнь Помпе, для лечения которых используют рекомбинантные препараты лизосомных ферментов. Однако редкие наследственные заболевания обычно диагностируют поздно, что не позволяет своевременно начать лечение и может негативно отразиться на его результатах. Генетическое тестирование родственников пробанда (семейный скрининг) дает возможность выявить наследственные болезни в более молодом возрасте, в том числе при отсутствии симптомов.
Болезнь Фабри (БФ) – X-сцепленная болезнь накопления, вызванная мутациями гена GLA, который кодирует лизосомный фермент α-галактозидазу. Дефицит активности этого фермента сопровождается прогрессирующим накоплением гликосфин голипидов, главным образом глоботриаозил церамида (GL3) и его деацилированной формы глоботриазилсфингозина (lyso-GL3), практически во всех клетках организма. Накопление гликосфинголипидов приводит к появлению различных симптомов, в том числе нейропатической боли, ангиокератом, желудочно-кишечных нарушений, нарушения потоотделения, непереносимости тепла/холода, а позднее вызывает поврежде ние органов-мишеней, в том числе почек, сердца и головного мозга [4-7]. Помимо классической БФ, описан ряд ее вариантов с более поздним, преимущественно сердечным фенотипом, связанных с такими мутациями, как p.F113L, p.N215S, IVS4+919G>A.
Роль семейного скрининга в диагностике болезни Фабри
Диагноз БФ часто устанавливают спустя много лет, а иногда десятилетий после появления первых симптомов [11,12]. С целью ранней диагностики БФ в некоторых странах, в том числе в Италии, Японии и Тайване, про водился скрининг среди новорожденных [13-16]. В этих исследованиях частота патогенных мутаций гена GLA составляла 0,03–0,08%. Кроме того, скрининг возможен в группах риска, например, у пациентов с гипертрофи ческой кардиомиопатией или больных, находящихся на гемодиализе, у которых вероятность наличия БФ значи тельно выше, чем в общей популяции. По данным мета-анализа более 60 исследований, мутации гена GLA были обнаружены, соответственно, у 0,21% и 0,15% мужчин и женщин, получавших лечение программным диализом, 0,94% и 0,90% – с гипертрофией левого желудочка неясного происхождения и 0,13% и 0,14% – перенесших инсульт в раннем возрасте [17]. В Российской Федерации общенациональный скрининг в диализных отделениях позволил выявить около полови ны из 78 пробандов с БФ, обследованных в клинике им. Е.М. Тареева [18]. Количество описанных мутаций гена GLA постоянно увеличивается и в настоящее время превышает 1000. В скрининговых исследованиях могут быть выявлены новые мутации, клиническое значение которых еще предстоит установить [17,19,20]. В 63 опубликованных скрининговых исследованиях, прово дившихся в группах риска, выявленные варианты гена GLA были непатогенными у 47,9% мужчин и 74,1% женщин [17]. В современных рекомендациях по веде нию пациентов с БФ указано, что при наличии вариан та гена GLA неизвестного значения необходимо проводить дополнительное обследование больных, включая анализ биоптатов, например, почки, для под тверждения патогенности мутации [21]. Ферменто заместительная терапия (ФЗТ) возможна только в тех случаях, когда диагноз БФ не вызывает сомнения и подтверждается не только результатами молекулярно генетического исследования, но и другими данными (активность a-галактозидазы А, клинические проявления, наличие заболевания у родственников и/или данные гистологического исследования).
В этом контексте составление подробной родословной пробанда с БФ в сочетании с семейным скринингом представляет собой мощный инструмент дляулучшения диагностики заболевания, в том числе ранней, а также определения клинического значенияновых мутаций GLA, выявленных в ходе различныхскрининговых программ. В одном исследовании БФбыла диагностирована в среднем у 5 членов семьи 74 пробандов с БФ [22], а в другом исследовании (31 родословная) на одного пробанда приходилось 15 больныхродственников [23]. В бразильском исследовании водной семье было выявлено 18 пациентов с БФ [24]. Вроссийской популяции средний показатель был ниже исоставил около 2 дополнительных случаев БФ в семьена одного пробанда, однако БФ была диагностированапочти у половины из 292 обследованных членов семейиндексных пациентов [25]. В последних рекомендацияхотражена важность клинического и генетического скрининга членов семей пациентов с впервые диагностированной БФ [21,26].
В 2020 г. группой экспертов, включая авторов данной статьи, был проведен систематизированный обзор публикаций в базах данных Embase и NCBI PubMed с целью поиска данных, касающихся семейного генетического анализа при БФ [1]. Основной целью этого исследования было оценить значение составления родословной и проведения семейного скрининга в диагностике БФ. В окончательный обзор вошли 89 публикаций, в том числе 49 сообщений о случаях, 36 клинических исследований, 3 обзорных статьи и 1 консенсус. В 82 из 89 публикаций сообщалось о 365 пробандах с БФ. При семейном скрининге БФ была диагностирована у 1744 членов семей индексных пациентов (в среднем 4,8 на одного пробанда). Хотя 65% пробандов составляли мужчины, почти две трети (65%) больных родственников, выявленных в результате генетического анализа, были женщинами. Семейный скрининг должен служить дополнением к скринингу новорожденных или скринингу в группах риска, в том числе среди диализных пациентов, больных гипертрофической кардиомиопатией или инсультом, развившимся в раннем возрасте. Например, в одном исследовании БФ была диагностирована у 99 родственников 21 пациента, выявленного при скрининге среди больных с гипертрофией левого желудочка неясного происхождения [27].
Семейный скрининг редких генетических заболеваний дает хорошие результаты, однако существует много барьеров (экономических, географических, социальных, общественных и культурных), которые могут препятствовать составлению родословных и проведению семейного генетического скрининга в различных регионах мира.
Составление и анализ семейных родословных
Когда у пациента впервые диагностируют редкое наследственное заболевание, такое как БФ, клиницист или генетик должны составить его подробную семейную родословную и идентифицировать членов семьи, которые могут быть иметь мутантный ген с учетом типа наследования (Х-сцепленного в случае БФ). Следует учитывать, что отсутствие симптомов, например, у детей или подростков или женщин, не исключает диагноз БФ, поэтому генетическое тестирование необходимо по возможности проводить всем родственникам больного, у которых потенциально можно выявить ту же мутацию гена GLA. При анализе родословной целесообразно также определить родственников, у которых генетическое исследование не имеет смысла. Например, при БФ мутантный ген, расположенный на Х-хромосоме, не передается от отца сыну.
Подробная семейная родословная при БФ может показаться слишком громоздкой и запутанной для неподготовленного специалиста. Однако после некоторой тренировки и изучения всех символов и примечаний семейная родословная станет идеальным способом, чтобы зафиксировать данные семейного анамнеза в упорядоченной и легкодоступной форме. Составление семейной родословной позволяет врачам легко визуализировать типы наследования или кластеризацию симптомов, что облегчает интерпретацию клинических фенотипов. В идеале семейные родословные следует создавать и хранить в цифровом виде, поскольку в этом случае их легче изменять и обновлять с течением времени. В настоящее время имеется несколько онлайн-приложений для составления родословных (например, http://www.apbenson.com/why-use-cyrillic/; http://pedigreedraw. com/; https://www.invitae.com/en/familyhistory/; https://phenotips.org/; http://www.progenygenetics.com/). Хотя эти приложения полезны, некоторые из них довольно сложны, поэтому сохраняется потребность в новых, удобных для пользователя и простых программных инструментах для составления родословных.
На рис. 1 представлен типичный пример семейной родословной, охватывающей 3 поколения, при БФ [1]. У 27-летнего мужчины (пробанд, III-1) без видимых причин развился инсульт. Была заподозрена и впоследствии диагностирована БФ (мутация p.Leu243Phe в GLA). Та же мутация была выявлена у его матери (II-2), а также брата, находившегося на лечении гемодиализом (III-3), и его дочери (IV-1). Родителя матери пробанда умерли (I-1 и I-2). Позднее диагноз БФ был установлен еще у 5 родственников пробанда по линии матери.
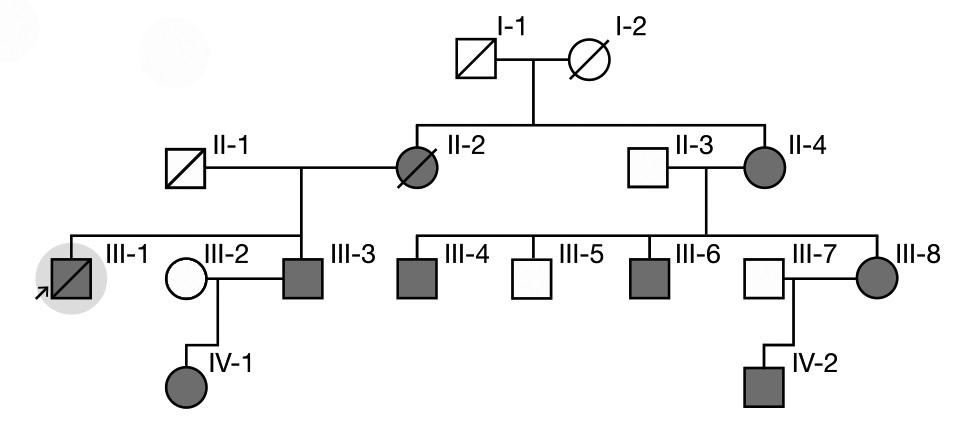
Существует несколько ключевых вопросов, которые необходимо учитывать при интерпретации генетической и клинической информации, полученной при анализе семейной родословной. Клинический фенотип у гетерозиготных по мутации гена GLA женщин зависит от типа мутации (миссенс или другие) и типа инактивации Х-хромосомы. У женщин имеются две Х-хромосомы, однако одна из них инактивируется во время раннего эмбрионального развития. В связи с этим для женщин характерен мозаицизм, т.е. наличие двух типов клеток, экспрессирующих материнскую или отцовскую Х-хромосому. При “случайной" инактивации Х-хромосом соотношение таких клеток составляет примерно 50:50, однако при “смещении" этого процесса доля клеток, экспрессирующих определенную Х-хромосому, может значительно увеличиться, что влияет на клинические проявления, тяжесть и прогрессирование заболевания у женщин с классической БФ [28]. Следовательно, женщины с одной и той же мутацией GLA могут иметь различные характер и степень тяжести симптомов, что усложняет интерпретацию семейного анамнеза и подчеркивает важность генотипирования всех потенциально затронутых членов семьи женского пола, независимо от наличия у них симптомов.
Еще один момент, о котором следует помнить при составлении родословной, это возможность умышленного или непреднамеренного утаивания истинной информации об отцовстве. Кроме того, в некоторых странах могут преобладать родственные браки, что может привести к появлению гомозиготных женщин. БФ может также развиться в результате редких (5–10% случаев) мутаций de novo [29].
Препятствия для проведения семейного скрининга и анализа родословных
Существует много факторов, препятствующих проведению или ограничивающих возможности семейного генетического скрининга на БФ (табл. 1) [1]. Они отличаются в разных регионах и странах в зависимости от стоимости скрининга и лечения, нормативно-правовых актов и т.д.
| Затраты |
| • Генетические тесты |
| • Консультация врача-генетика |
| • Транспортировка образцов высушенной крови |
| • Дорожные расходы для пациента |
| Культурные/общественные проблемы |
| • Сложные структуры семей |
| • Разобщенные семьи |
| • Кровнородственные браки/эндогамия |
| • Патриархальные общества |
| • Боязнь стигматизации |
| Логистические проблемы |
| Географическое разобщение населения (низкая плотность населения, удаленные районы) |
| • Внутренняя миграция семей, затрудняющая контакты |
| • Слабая национальная инфраструктура |
| • Нехватка квалифицированных врачей-генетиков |
| Коммуникации |
| • Плохая осведомленность врачей о БФ и пользе скрининга |
| • Низкий уровень образования пациентов, затрудняющий общение с ними |
| • Трудности с отслеживанием родственников |
Стоимость молекулярно-генетического исследования может препятствовать проведению семейного скрининга во многих странах, хотя эти затраты следует соотносить со стоимостью лечения потенциально предотвратимых осложнений заболевания, таких как прогрессирующая почечная недостаточность или инсульт. В развивающихся странах с обширными малонаселенными регионами важным финансовым барьером может быть стоимость консультаций генетиков. В некоторых странах, в том числе в России, помощь в покрытии расходов на семейный скрининг оказывают организации пациентов. В Индии, где отсутствует национальная политика в отношении ФЗТ при БФ, существуют благотворительные программы, помогающие пациентам компенсировать затраты на диагностику и лечение [1].
Существует много культурных и социальных факторов, влияющих на возможность проведения семейного генетического анализа при редких заболеваниях. В некоторых географических регионах, например, в Омане, поощряются близкородственные браки, несмотря на известный риск увеличения распространенности генетических заболеваний, в частности, аутосомнорецессивных, таких как спинальная мышечная атрофия, муковисцидоз, фенилкетонурия, рецессивно наследуемые врожденные пороки развития и умственная отсталость [30]. Поскольку БФ имеет X-сцепленный тип наследования, близкородственные браки должны увеличивать распространенность гомозиготной формы болезни у женщин, у которых, как ожидается, будут проявляться более тяжелые клинические фенотипы. В связи с возможной стигматизацией необходимо уделять особое внимание сохранению конфиденциальности данных при общении с пациентами и членами их семей. Сложные семейные структуры, существующие в некоторых странах, например, в Мексике, где мужчины могут иметь две или три семьи, также затрудняют семейный скрининг и анализ родословных [1]. Во многих странах и регионах члены семей могут быть разобщены географически и не контактировать друг с другом, что затрудняет отслеживание родственных связей пациента с впервые выявленной БФ. В некоторых странах эта проблема связана с высоким уровнем внутренней миграции или региональными конфликтами. В других странах, например, в Саудовской Аравии, существуют весьма консервативные, патриархальные общества, в которых мужья/отцы препятствуют проведению генетического анализа среди женщин [1]. Такие глубоко укоренившиеся культурные и религиозные предрассудки бывает крайне трудно преодолеть. В некоторых развивающихся странах, например в Индии, существует значительная стигматизация, связанная с наличием в семье пациентов с диагнозом редкого наследственного заболевания, которые не могут получить доступ к дорогостоящему лечению. Некоторые пациенты часто предпочитают жить в надежде на то, что не страдают каким-либо заболеванием, включая БФ, и не пытаются пройти генетический анализ. Хотя в Южной Корее ФЗТ при БФ полностью финансируется государством с 2003 г., уровень диагностики этого заболевания до сих пор остается достаточно низким. Авторы недавно опубликованного общенационального опроса отчасти объясняют это отказом от семейного генетического анализа из-за страха стигматизации [31]. Религиозные убеждения могут также создавать препятствия для семейного скрининга, что имеет место в некоторых общинах в Мексике и во многих странах с консервативным или сильно религиозным населением.
Географическое распределение населения на больших пространствах, включая труднодоступные районы, может создать логистические проблемы для реализации программ семейного скрининга БФ в некоторых странах, например, в России, Китае и Колумбии. Семьи могут мигрировать на большие расстояния, что затрудняет контакты между их членами. Кроме того, многие развивающиеся страны имеют слабую коммуникационную инфраструктуру и плохую транспортную сеть, что усугубляет проблемы диагностики в тех случаях, когда региональные генетические службы немногочисленны или вообще отсутствуют. Пациентам, как правило, приходится преодолевать большие расстояния до столицы, чтобы пройти диагностику и получить помощь специалиста. В более обеспеченных странах образцы высушенных пятен крови, требуемые для генетического анализа, могут быть доставлены курьером в генетическую лабораторию. Например, в России существует финансируемая курьерская служба, которая позволяет медицинским работникам в любом городе бесплатно отправить высушенные пятна крови в генетическую лабораторию. Тем не менее, в России необходимо разработать и утвердить национальные нормативно-правовые акты, регулирующие проведение генетического анализа, а также зарегистрировать соответствующие реактивы и диагностическое оборудование.
В развивающихся странах низкие уровень образования и доход на душу населения могут быть серьезными препятствиями для диагностики наследственных заболеваний, семейного скрининга и доступа к лечению. Многие пациенты не имеют официального трудоустройства, номеров социального страхования и/или медицинской страховки и, следовательно, не могут получить лечение, которое предоставляется государственными медицинскими службами. Низкий уровень образования, особенно в сельских районах, может создавать проблемы для врачей при разъяснении пациентам и их родственникам важности диагностики редкого наследственного заболевания и семейного скрининга. Трудности в общении часто еще более усугубляются религиозными аспектами, которые часто выдвигаются в качестве причин, по которым не следует проводить генетический анализ. Поэтому важен поиск стратегий, помогающих врачам общаться с новыми пациентами, а пациентам – с членами своих семей. Во многих случаях пациенты, у которых было впервые выявлено то или другое заболевание, плохо представляют, что это для них означает. Простая брошюра или буклет, содержащие ключевые факты о БФ, помогут информировать пациента. Наличие стандартного набора вопросов может быть полезным для пациентов, когда они обсуждают свой диагноз со своими родственниками. Социальные сети могут играть определенную роль в поиске родственников и восстановлении контактов. Кроме того, в зависимости от региона/страны, может потребоваться просветительская работа среди врачей общей практики о важности генетического анализа на редкие наследственные заболевания.
Как можно улучшить семейный скрининг при болезни Фабри
Значительную помощь в организации семейного скрининга могут оказать организации пациентов. Например, в Аргентине имеется около 700 пациентов с БФ, контакты с которыми затруднены из-за плохого охвата страны сетями мобильной телефонной связи и интернет. В стране существуют две национальные ассоциации пациентов с БФ, которые ежегодно проводят одну общую конференцию [1]. Также в Аргентине есть два генетических консультанта, оказывающих поддержку пациентам с БФ. Сами больные также принимают активное участие в организации семейного скрининга. Они убеждают своих родственников пройти генетический анализ, а также каждый год заполняют анкеты, помогающие выявить новые случаи в своих семьях. Очевидно, что хорошо информированные пациенты, которые имеют представления о своем заболевании и типе ее наследования, мотивированы к поиску новых членов семьи, которым могут помочь ранняя диагностика и лечение.
Почки часто поражаются при БФ, а нефрологи играют важную роль в диагностике этого заболевания. Скрининг на БФ целесообразно проводить среди мужчин младше 50 лет и женщин любого возраста с хронической болезнью почек (ХБП) неизвестной этиологии, особенно при наличии других возможных проявлений заболевания [32]. Технически относительно несложно провести скрининг в диализных центрах, которые пациенты с терминальной стадией хронической почечной недостаточности посещают три раза в неделю. В Бразилии проведено скрининговое исследование среди 2583 мужчин, находящихся на гемодиализе в 23 центрах [24]. БФ была диагностирована у 3 пациентов (0,12%). В ходе последующего семейного скрининга диагноз БФ был подтвержден у 5 родственников первого пробанда, 18 родственников второго и 2 родственников третьего. Анализ родословной помог также подтвердить патогенность новой миссенс-мутации (p.C52F), обнаруженной у третьего пробанда. В другом скрининговом исследовании, проведенном в одном бразильском диализном центре [33], среди 108 мужчин, находившихся на гемодиализе, был выявлен 1 пациент с БФ (0,9%). У этого пациента определялись новая миссенс-мутация (p.G35V) и классический фенотип БФ. При семейном скрининге были выявлены еще 9 членов семьи с БФ. Эти исследования подчеркивают важность семейного скрининга как инструмента для выявления пациентов с БФ в более раннем возрасте, когда эффективность ФЗТ может быть выше, а также для оценки патогенности новых мутаций. В России значительную часть выявленных пациентов с БФ составляют больные, находящиеся на гемодиализе (n=44), у которых диагноз был установлен в процессе общенационального скрининга в диализных отделениях, и члены их семей [34].
В современных рекомендациях указывается, что всех пациентов с впервые диагностированной БФ следует направлять на генетическое консультирование для составления семейной родословной и планирования семейного скрининга [26]. Однако во многих странах генетические консультанты недоступны или не имеют юридического статуса медицинских работников. Например, население Колумбии составляет более 48 миллионов человек, а плотность населения – 42 человека на км2. Диагностика БФ в Колумбии затруднительна, так как в стране мало врачей-генетиков, а в столице имеется только одна лаборатория [1]. Однако в стране есть специалист, спонсируемый государственной службой здравоохранения, который может совершать поездки к пациентам и/или их родственникам для оказания необходимой помощи в диагностике заболевания и проведении семейного скрининга. При этом ему не разрешается участвовать в процессе принятия решения о лечении, которое принимается вторым врачом. В настоящее время в Колумбии зарегистрировано 20 индексных случаев БФ, а 18 семей согласились обратиться за помощью к специалисту по семейному скринингу.
Мексике частота выявления БФ низкая, а в стране нет генетических консультантов. С целью улучшения диагностики заболевания в 2015 г. была организована специальная программа семейного скрининга, включающая составление родословных, в которой задействованы более 10 генетиков и пациенты из 17 штатов страны [1]. В рамках программы проводятся регулярные встречи для обсуждения практического опыта, препятствий или проблем, с которыми сталкиваются генетики, а также разработки стратегий улучшения программы.
На сегодняшний день в программе приняли участие 40 семей, в результате чего было выявлено 93 новых пациента с БФ [1]. Помимо нехватки генетических консультантов существуют и другие препятствия для семейного скрининга в Мексике, в том числе сложные семейные структуры, низкий уровень образования среди некоторых подгрупп населения и широкая рассредоточенность сельских жителей. В целом мексиканские врачи мало осведомлены о БФ, а многие даже не желают знать, диагностировать и лечить ее. Кроме того, в Мексике широко распространены хронические дегенеративные заболевания, что повышает вероятность ошибочной диагностики БФ.
Важность ранней диагностики для повышения эффективности лечения
Своевременная диагностика редкого наследственного заболевания, которое поддается лечению, очень важна, поскольку раннее назначение патогенетической терапии может улучшить ее результаты. Лечение БФ предполагает применение рекомбинантных препаратов α-галактозидазы – агалсидазы альфа (0,2 мг/кг) или агалсидазы бета (1 мг/кг), которые вводят внутривенно каждые 2 недели. За рубежом одобрен для применения мигаластат, который представляет собой шаперон, повышающий остаточную активность фермента. Этот препарат используется для лечения пациентов с определенными мутациями гена GLA. В нескольких крупных исследованиях установлено, что более ранняя ФЗТ позволяет добиться большего эффекта по сравнению с таковым у больных, которые начинают лечение на более позднем этапе, когда имеется необратимое повреждение органов [35,36].
Заключение
Генетический анализ в семьях индексных пациентов, у которых заболевание было выявлено на основании имеющихся симптомов или в рамках скрининговой программы, может значительно увеличить количество диагностированных случаев редких наследственных заболеваний и облегчить их раннюю диагностику. Это подтверждается результатами семейного скрининга при БФ, который в опубликованных исследованиях позволил выявить в среднем еще около 5 пациентов на одного пробанда (в России этот показатель был ниже и составил около 2 на одного индексного пациента). Семейный скрининг дает также возможность диагностировать заболевание в более молодом возрасте, в том числе при отсутствии клинических симптомов, и начать лечение на раннем этапе, когда отсутствуют необратимые изменения внутренних органов. Изучение родословных помогает определить патогенность новых мутаций, которые не были описаны ранее. Важное значение для повышения эффективности семейного скрининга имеют создание более простых программных средств для построения родословных и подготовка образовательных материалов для пациентов.
Используемые источники
- Germain DP, Moiseev S, Suarez-Obando F, et al. The benefits and challenges of family genetic testing in rare genetic diseases – lessons from Fabry disease. Mol Genet Genomic Med. 2021;00:e1666.
- Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med 2003;138:338-46.
- Germain DP. Fabry disease. Orphan J Rare Dis 2010;5:30.
- Hagege A, Reant P, Habib G, et al. Fabry disease in cardiology practice: Literature review and expert point of view. Arch Cardiovasc Dis 2019;112:278-87.
- Kolodny E, Fellgiebel A, Hilz MJ, et al. Cerebrovascular involvement in Fabry disease: Current status of knowledge. Stroke 2015;46:302-13.
- Linhart A, Germain DP, Olivotto I, et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Europ J Heart Fail 2020;22:1076-96.
- Ortiz A, Cianciaruso B, Cizmarik M, et al. End-stage renal disease in patients with Fabry disease: Natural history data from the Fabry Registry. Nephrol Dial Transpl 2010;25:769-75.
- Oliveira JP, Nowak A, Barbey F, et al. Fabry disease caused by the GLA p.Phe113Leu (p.F113L) variant: Natural history in males. Europ J Med Gen 2020;63:103703.
- Germain DP, Brand E, Burlina A, et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol Gen Genom Med 2018;6:492-503.
- Hsu TR, Hung S, Chang FP, et al. Later onset Fabry disease, cardiac damage progress in silence: Experience with a highly prevalent mutation. J Amer Coll Cardiol 2016;68:2554-63.
- Mehta A, Beck M, Eyskens F, et al. Fabry disease: A review of current management strategies. QJM 2010;103:641-59.
- Reisin R, Perrin A, GarcТa-PavТa P. Time delays in the diagnosis and treatment of Fabry disease. Intern J Clin Pract 2017;71:12914.
- Burlina AB, Polo G, Salviati L, et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J Inher Metab Dis 2018;41:209-19.
- Chinen Y, Nakamura S, Yoshida T, et al. A new mutation found in newborn screening for Fabry disease evaluated by plasma globotriaosylsphingosine levels. Human Genome Var 2017;4:17002.
- Liao HC, Hsu TR, Young L, et al. Functional and biological studies of alphagalactosidase A variants with uncertain significance from newborn screening in Taiwan. Mol Gen Metab 2018;123:40-147.
- Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Amer J Hum Genet 2006;79:31-40.
- Doheny D, Srinivasan R, Pagant S, et al. Fabry disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995-2017. J Med Genet 2018;55:261-8.
- Моисеев С.В., Тао Е.А., Моисеев А.С. и др. Клинические проявления и исходы болезни Фабри у 150 взрослых пациентов. Клин фармакол тер 2021;30(3):43-51 [Moiseev S, Tao E, Moiseev A, et al. Clinical manifestations and outcomes of Fabry disease in 150 adult patients. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(3):43-51 (In Russ.)].
- Varela P, Mastroianni Kirsztajn G, Motta FL, et al. Correlation between GLA variants and alpha-galactosidase A profile in dried blood spot: An observational study in Brazilian patients. Orphan J Rare Dis 2020;15:30.
- Germain DP, Oliveira JP, Bichet DG, et al. Use of a rare disease registry for establishing phenotypic classification of previously unassigned GLA variants: A consensus classification system by a multispecialty Fabry disease genotype-phenotype workgroup. J Med Genet 2020;57:542-51.
- Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet 2019;96:107-17.
- Laney DA, Germain DP, Oliveira JP, et al. Fabry disease and COVID-19: International expert recommendations for management based on real-world experience. Clin Kidney J 2020;13:913-25.
- Rozenfeld PA, Masllorens FM, Roa N, et al. Fabry pedigree analysis: A successful program for targeted genetic approach. Mol Gen Genom Med 2019;7:e00794.
- Silva CA, Barreto FC, Dos Reis MA, et al. Targeted screening of Fabry disease in male hemodialysis patients in Brazil s importance of family screening. Nephron 2016;134:221-30.
- Тао Е.А., Моисеев А.С., Новиков П.И. и др. Эффективность семейного скрининга при болезни Фабри в Российcской популяции. Клин фармакол тер 2020;29(2):34-9 [Tao E, Moiseev A, Novikov P, et al. Efficacy of family screening in Fabry disease in the Russian population. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2020;29(2):34-9 (In Russ.)].
- Ortiz A, Germain DP, Desnic Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab 2018;123:416-27.
- Azevedo O, Gal A, Faria R, et al. Founder effect of Fabry disease due to p.F113L mutation: Clinical profile of a late-onset phenotype. Mol Genet Metab 2020; 129:150-60.
- Echevarria L, Benistan K, Toussaint A, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet 2016;89:44-54.
- Gal A, Beck M, HЪppner W, Germain DP. Clinical utility gene card for Fabry disease - update 2016. Europ J Hum Genet 2017;25:e1-3.
- Rajab A, Al Rashdi I, Al Salmi Q. Genetic services and testing in the Sultanate of Oman. Sultanate of Oman steps into modern genetics. J Commun Gen 2013;4:391-7.
- Choi JH, Lee BH, Heo SH, et al. Clinical characteristics and mutation spectrum of GLA in Korean patients with Fabry disease by a nationwide survey: Underdiagnosis of late-onset phenotype. Medicine 2017;96:e7387.
- Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: Conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Intern 2017;91:284-93.
- Veloso V, Ataides TL, Canziani M, et al. A novel missense GLA mutation (p. G35V) detected in hemodialysis screening leads to severe systemic manifestations of Fabry disease in men and women. Nephron 2018;138:147-56.
- Moiseev S, Fomin V, Savostyanov K, et al. The prevalence and clinical features of Fabry disease in hemodialysis patients: Russian Nationwide Fabry Dialysis Screening Program. Nephron 2019;141(4):249-55.
- Germain DP, Elliott PM, Falissard B, et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: A systematic literature review by a European panel of experts. Mol Genet Metab Rep 2019;19:100454.
- Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab 2108;124:189-203.