Клинико-рентгенологическая характеристика интерстициального поражения легких при первичном билиарном холангите
Изучить частоту, особенности клиникорентгенологической картины и факторы риска интерстициального поражения легких (ИПЛ) при первичном билиарном холангите (ПБХ).
В проспективное когортное исследование были включены 93 пациента (89 женщин, средний возраст на момент дебюта заболевания – 47,1±1,0 лет) с ПБХ без сопутствующих системных заболеваний соединительной ткани. Всем пациентам проводили компьютерную томографию органов грудной клетки и исследование функции внешнего дыхания. Оценивали легочную и печеночную выживаемость. Неблагоприятным легочным исходом считали смерть от осложнений ИПЛ, комбинированным неблагоприятным печеночным исходом – смерть от осложнений цирроза печени или трансплантацию печени. Медиана длительности наблюдения составила 29 (11,5; 38,0) мес.
Различные варианты ИПЛ были диагностированы у 38 (40,9%) из 93 пациентов. Среди них чаще всего встречались саркоидоподобное ИПЛ, субклиническое ИПЛ и организующаяся пневмония. Группа пациентов с ИПЛ характеризовалась меньшей долей больных c циррозом печени и симптомами заболевания печени, а также более высоким средним уровнем IgM сыворотки крови и большей частотой антимитохондриальных антител М2-фракции (АМА-М2). При многофакторном анализе отсутствие печеночных симптомов в дебюте заболевания (отношение шансов [ОШ] 11,5; 95% доверительный интервал [ДИ] 1,2-109,4; p=0,033), наличие печеночных неказеозных эпителиоидноклеточных гранулем (ОШ 17,8; 95% ДИ 1,8-174,6; p=0,014), более высокие титры IgM сыворотки крови (ОШ 1,5; 95% ДИ 1,1-2,2; p=0,020) и лейкоцитов крови (ОШ 2,4; 95% ДИ 1,2-4,7; p=0,016) были независимыми факторами, ассоциировавшимися с наличием ИПЛ при ПБХ. Более трети пациентов с ИПЛ не имели каких-либо респираторных симптомов, и только один умер от осложнений ИПЛ в течение периода наблюдения. Пациенты с ИПЛ имели лучшую печеночную выживаемость.
Результаты нашего исследования продемонстрировали высокую распространенность ИПЛ, в том числе бессимптомного, среди пациентов с ПБХ. В большинстве наблюдений ПБХ-ассоциированное ИПЛ имело доброкачественное течение, не определяло жизненный прогноз больного и ассоциировалось с меньшей тяжестью поражения печени. ИПЛ при ПБХ следует включать в дифференциальный диагноз интерстициальных заболеваний легких.
Первичный билиарный холангит (ПБХ) – аутоиммунное холестатическое заболевание печени, возникающее вследствие иммуноопосредованного поражения мелких внутрипеченочных желчевыводящих протоков и приводящее к циррозу печени. Морфологическим субстратом заболевания является хронический негнойный деструктивный холангит, вовлекающий междольковые желчевыводящие протоки. Другой характерный гистологический признак ПБХ – эпителиоидноклеточные гранулемы без некроза, выявляемые в печени у части больных преимущественно на ранних стадиях заболевания [1]. При ПБХ в сыворотке крови определяются антимитохондриальные антитела М2 фракции (АМА-М2), различные антинуклеарные антитела (АНА) и повышение сывороточного уровня иммуноглобулина М (IgM). Заболевание поражает преимущественно женщин в возрасте от 40 до 50 лет и клинически проявляется кожным зудом, гиперпигментацией кожи, желтухой и гепато спленомегалией [1]. Препарат выбора при лечении ПБХ – урсодезоксихолевая кислота (УДХК), применение которой позволяет уменьшить клинико-лабораторные проявления холестаза, замедлить прогрессирование заболевания и уменьшить риск наступления неблагоприятного печеночного исхода [1,2]. К средствам второй линии у пациентов, не ответивших на лечение УДХК, относят фибраты и обетихолевую кислоту [2].
ПБХ свойственно наличие разнообразных внепеченочных иммунных проявлений, таких как аутоиммунный тиреоидит, синдром Шегрена и хронический тубулоинтерстициальный нефрит [3-5]. К более редким системным проявлениям ПБХ относят различные варианты интерстициального поражения легких (ИПЛ), в том числе организующуюся пневмонию (ОП) [6,7], лимфоцитарную интерстициальную пневмонию [8,9], неспецифическую интерстициальную пневмонию (НСИП) [10,11], обычную интерстициальную пневмонию (ОИП) [12], легочный саркоидоподобный гранулематоз [13]. Продемонстрирована возможность развития иммунного альвеолита у пациентов с ПБХ, не имеющих ИПЛ [14-16]. Первоначально предполагалось, что как ИПЛ, так и поражение дыхательных путей у пациентов с ПБХ объясняется легочными проявлениями сопутствующего синдрома Шегрена [17,18], однако поражение легких было описано и у пациентов с ПБХ, не имевших каких-либо системных заболеваний соединительной ткани (СЗСТ) [19,20].
В настоящее время остаются недостаточно изученными распространенность различных вариантов ИПЛ при ПБХ, а также их ключевые характеристики. Неизвестно, существует ли связь между наличием ПБХ-специфичных АНА и формированием ИПЛ у носителей данных антител. Целью исследования была оценка частоты, особенностей клинико-рентгенологической картины и факторов риска развития ИПЛ при ПБХ.
Материал и методы
Проспективное когортное исследование проводили с января 2018 до января 2022 года на базе клиники им. Е.М. Тареева Университетской клинической больницы №3 Первого МГМУ им. И.М. Сеченова. Диагноз ПБХ устанавливали на основании критериев Американской ассоциации по изучению заболеваний печени [2]. Сочетание ПБХ с аутоимунным гепатитом (ПБХ-АИГ) диагностировали на основании Парижских критериев [21]. Биохимический ответ на лечение УХДК оценивали согласно критериям, предложенным С. Corpechot и соавт. [22]. В исследование не включали пациентов, имевших наряду с ПБХ хронический вирусный гепатит В и С или СЗСТ, в том числе синдром Шегрена, системную склеродермию, дерматомиозит или полимиозит, ревматоидный артрит, системную красную волчанку и IgG4-ассоциированное заболевание.
В ходе исследования применяли стандартные клинические, лабораторные и инструментальные методы обследования больных гепатологического профиля. Дополнительно исследовали панель АНА аутоиммунных заболеваний печени (EuroImmune, Германия) методом иммуноферментного анализа у 46 пациентов. Всем пациентам, принимавшим участие в исследовании, проводили компьютерную томографию (КТ) органов грудной клетки высокого разрешения и спирометрию. При спирометрии оценивали значения форсированной жизненной емкости легких (ФЖЕЛ) и объем форсированного выдоха за 1 с (ОФВ1) в процентах от должных величин [23]. КТ-паттерны ИПЛ классифицировали согласно рекомендациям Американского торакального общества и Европейского респираторного общества [24]. Двусторонние ретикуло-нодулярные измененения с перилимфатическим распределением изолированно или в сочетании с двусторонним увеличением лимфатических узлов средостения классифицировали как саркоидоподобный паттерн ИПЛ. Неклассифицируемый вариант ИПЛ диагностировали при невозможности отнести выявленные рентгенологические и гистологические интерстициальные изменения к определенному паттерну ИПЛ. Наличие так называемого "субклинического" ИПЛ (subclinical ILD) констатировали при выявлении интерстициальных изменений в легких, занимавших менее 5% от общей площади легочной паренхимы, отсутствии любых респираторных симптомов и сохранной вентиляционной функции легких (ФЖЕЛ >80% от должной).
Плотность печени оценивали методом эластографии на аппарате "Фиброскан" (Echosense, Франция) у 35 пациентов. Гистологическое исследование биоптатов печени проводили у 45 пациентов, биоптатов легкого – у 8, внутригрудных и надключичных лимфатических узлов – у 4.
Статистическую обработку результатов исследования проводили с помощью программного пакета SPSS версии 22.0. Нормальность распределения количественных признаков определяли с помощью критерия Шапиро-Уилка. Переменные в зависимости от нормальности распределения представлены в виде средних значений и стандартных отклонений или медиан и межквартильного интервала. Качественные переменные приведены в виде абсолютных значений и доли в процентах. При нормальном распределении количественные показатели двух независимых групп сравнивали с помощью критерия Стьюдента, при распределении, отличающемся от нормального, – с помощью критерия Манна-Уитни. Сравнение групп по качественным признакам проводилось с помощью критерия χ2 Пирсона и точного критерия Фишера при количестве наблюдений в группе менее 10. Для параметров, выявленных при однофакторном анализе, проводили многофакторный анализ методом бинарной логистической регрессии. При анализе выживаемости использовали метод Каплана-Майера, для оценки достоверности различий выживаемости между сравниваемыми группами применяли логарифмический ранговый критерий. Факторы, влияющие на риск наступления анализируемых исходов, определяли при помощи модели пропорциональных рисков Кокса. При анализе легочной выживаемости за исход принимали смерть вследствие осложнений ИПЛ. При анализе печеночной выживаемости в качестве комбинированного исхода принимали смерть от осложнений цирроза печени или трансплантацию печени.
Исследование было проведено в соответствии с этическими принципами медицинских исследований, изложенными в Хельсинкской декларации, и было одобрено локальным этическим комитетом.
Результаты
Характеристика больных. В исследование были включены 93 пациента, большинство из которых составили женщины (95,7%). Средний возраст на момент дебюта заболевания составил 47,1±1,0 лет. Другие клинические и демографические показатели изучаемой когорты представлены в табл. 1. Пациентов, участвовавших в исследовании, наблюдали в течение 29 (11,5; 38,0) мес. Средняя продолжительность наблюдения за больными, имевшими и не имевшими ИПЛ, значимо не отличалась и составила 31 и 25 месяцев, соответственно (р=0,122).
Более половины пациентов в изучаемой когорте имели цирроз печени, почти у каждого шестого было диагностировано сочетание ПБХ с АИГ. АМА-М2 были выявлены в сыворотке крови у 84 (90,3%) пациентов. Частота обнаружения различных ПБХ-специфических АНА приведена в табл. 1. Все пациенты получали лечение УДХК, тогда как больным с перекрестным синдромом ПБХ-АИГ дополнительно проводили иммуносупрессивную терапию системными глюкокортикостероидами в сочетании с азатиоприном.
Интерстициальное поражение легких. КТ-признаки ИПЛ были выявлены у 38 (40,9%) пациентов. У 14 (36,8%) из 38 пациентов определялось саркоидоподобное ИПЛ, у 9 (23,7%) – "субклиническое" ИПЛ, у 7 (18,4%) – ОП, у 4 (10,5%) – неклассифицируемое ИПЛ, у 3 (7,9%) – НСИП и у 1 (2,6%) – ОИП. У 4 пациентов с рентгенологической картиной, не соответствовавшей известным паттернам ИПЛ, наблюдали следующие изменения: множественные мелкие очаги со смешанным паттерном распределения (у 2), утолщение междольковых перегородок в сочетании с зонами матового стекла и единичными мелкими очагами в базальных отделах легких (у 1), двустороннее увеличение внутригрудных лимфатических узлов в сочетании с распространенными зонами матового стекла и фокусом с эффектом обратного гало (у 1).
Гистологическое исследование было проведено у 9 пациентов с саркоидоподобным КТ-паттерном (видеоассистированная торакоскопическая биопсия легкого у 5, трансбронхиальная биопсия лимфатических узлов средостения у 2, резекция надключичных лимфатических узлов у 2). Во всех случаях были обнаружены множественные неказеозные эпителиоидно-клеточные гранулемы, сливающиеся в конгломераты и замещавшие нормальную ткань. Хирургическая биопсия легкого, проведенная у 2 пациентов с КТ-паттерном ОП, показала наличие полиповидной грануляционной ткани, заполняющей просветы терминальных бронхиол и альвеол, что соответствовало рентгенологической картине. Таким образом, во всех вышеупомянутых случаях, результаты гистологического исследований подтвердили рентгенологический диагноз.
Гистологические изменения в ткани легких у 3 пациентов с неклассифицируемым КТ-паттерном включали плотную лимфоцитарную инфильтрацию межальвеолярных перегородок (n=3), единичные гигантские клетки (n=3), рыхлые неказеозные макрофагальные и эпителиоидно-клеточные гранулемы (n= 2), пенистые макрофаги (n=2), скопления эозинофилов (n=2) и умеренный интерстициальный фиброз (n=1). В данных наблюдениях результаты патоморфологического исследования подтвердили вовлечение легочного интерстиция, однако не согласовывались с КТ-картиной, вследствие чего классифицировать ИПЛ не представлялось возможным.
Обусловленные ИПЛ респираторные симптомы были первым клиническим проявлением ПБХ у 9 (9,6%) пациентов, однако у всех из них имелись отклонения лабораторных показателей функции печени на момент манифестации поражения легких. Наиболее распространенными клиническими проявлениями ИПЛ были хронический кашель без мокроты (n=19, 50,0%), лихорадка (n=17, 44,7%), одышка в покое и при физической нагрузке (n=16, 42,1%). Дыхательная недостаточность была диагностирована у 3 (7,9%) из 38 пациентов с ИПЛ. У 14 (36,8%) из 38 пациентов с ИПЛ отсутствовали какие-либо респираторные симптомы в течение периода наблюдения.
Группы пациентов с ИПЛ и без ИПЛ существенно не отличались по возрасту, половому составу, частоте курения и ПБХ-АИГ, показателям ОФВ1 и ФЖЕЛ, однако ожирение чаще встречалось у больных с ИПЛ, а цирроз печени – у пациентов без ИПЛ (табл. 1). Плотность печени была ниже у больных с ИПЛ (7,9 и 14,2 кПа, соответственно, р=0,006). Отсутствие симптомов заболевания печени в дебюте заболевания чаще отмечали у больных с ИПЛ.
| Показатели | Все пациенты (n=93) |
Пациенты с ИПЛ (n=38) |
Пациенты без ИПЛ (n=55) |
p |
|---|---|---|---|---|
| Женщины, n (%) | 89 (95.7%) | 37 (97.4%) | 52 (94.5%) | 0,642 |
| Возраст начала заболевания, лет | 47,1±1,0 | 45,4±10,4 | 48,2±9,5 | 0,181 |
| Курение, n (%) | 18 (19,4) | 7 (18,4) | 11 (20,0) | 1,000 |
| Ожирение, n (%) | 21 (22,6) | 13 (34,2) | 8 (14,5) | 0,042 |
| ПБХ-АИГ, n (%) | 16 (17,2) | 9 (23,7) | 7 (12,7) | 0,263 |
| Цирроз печени, n (%) | 49 (52,7) | 15 (39,5) | 34 (61,8) | 0,034 |
| Гранулемы печени, n (%) | 11/45 (24,4) | 8/18 (44,4) | 3/27 (11,1) | 0,016 |
| Плотность печени, кПА | 11,4 (6,9; 17,4) | 7,9 (5,6; 12,6) | 14,2 (8,2; 26,3) | 0,006 |
| ОФВ1, % от должного | 102,0 (88,7; 115,7) | 99,5 (90,0; 117,2) | 105,0 (88,0; 110,7) | 0,807 |
| ФЖЕЛ, % от должного | 101,0 (90,0; 110,0) | 99,0 (85,0; 114,3) | 103,0 (91,5; 110,0) | 0,373 |
| Отсутствие печеночных симптомов в дебюте болезни, n (%) | 33 (35,5) | 20 (52,6) | 13 (23,6) | 0,004 |
| Кожный зуд, n (%) | 63 (67,7) | 20 (52,6) | 43 (78,2) | 0,010 |
| Гиперпигментация кожи, n (%) | 37 (39,8) | 8 (21,1) | 29 (52,7) | 0,002 |
| Асцит, n (%) | 16 (17,2) | 4 (10,5) | 12 (21,8) | 0,176 |
| Варикозное расширение вен пищевода, n (%) | 36 (38,7) | 13 (34,2) | 23 (41,8) | 0,459 |
| Печеночная энцефалопатия, n (%) | 11 (11,8) | 2 (5,3) | 9 (16,4) | 0,190 |
| Сухой синдром, n (%) | 34 (36,6) | 13 (34,2) | 21 (38,2) | 0,696 |
| Суставной синдром, n (%) | 33 (35,5) | 17 (44,7) | 16 (29,1) | 0,121 |
| Суставной синдром, n (%) | 33 (35,5) | 17 (44,7) | 16 (29,1) | 0,121 |
| Лихорадка, n (%) | 21 (22,6) | 17 (44,7) | 4 (7,3) | <0,001 |
| Одышка, n (%) | 22 (23,7) | 16 (42,1) | 6 (10,9) | 0,001 |
| Кашель, n (%) | 25 (26,9) | 19 (50,0) | 6 (10,9) | <0,001 |
| Тромбоциты, ×109/л | 194,6±9,9 | 215,7±81,7 | 180,4±102,5 | 0,083 |
| Лейкоциты, ×109/л | 5,2 (3,8; 6,7) | 6,0 (4,6; 7,4) | 4,4 (3,4; 6,0) | 0,001 |
| Нейтрофилы, ×109/л | 2,8 (2,0; 3,7) | 3,2 (2,6; 4,5) | 2,5 (2,0; 3,1) | 0,003 |
| Альбумин, г/л | 40,6 (36,0; 42,6) | 40,3 (36,4; 42,0) | 40,6 (35,3; 42,8) | 0,827 |
| Альбумин, г/л | 40,6 (36,0; 42,6) | 40,3 (36,4; 42,0) | 40,6 (35,3; 42,8) | 0,827 |
| Общий билирубин, ммоль/л | 21,4 (13,1; 39,5) | 16,7 (12,5; 30,8) | 23,2 (15,5; 46,4) | 0,084 |
| Щелочная фосфатаза, ЕД/л | 507,0 (296,5; 1018,7) | 585,0 (230,5; 1053,0) | 487,0 (319,0; 945,0) | 0,914 |
| Гамма-глутамилтранспептидаза, ЕД/л | 128,5 (45,0; 339,0) | 148,0 (42,5; 463,0) | 124,0 (48,0; 255,0) | 0,602 |
| IgM, г/л | 2,6 (1,8; 4,2) | 3,1 (2,1; 6,2) | 2,3 (1,7; 3,4) | 0,033 |
| Ревматоидный фактор, n (%), | 25/75 (33,3) | 13/32 (40,6) | 12/43 (27,9) | 0,248 |
| Ревматоидный фактор, n (%), | 25/75 (33,3) | 13/32 (40,6) | 12/43 (27,9) | 0,248 |
| AMA-M2, n (%) | 84 (90,3) | 38 (100) | 46 (83,6) | 0,010 |
| Анти-gp210, n (%) | 22/46 (47,8) | 5/14 (35,7) | 17/32 (53,1) | 0,346 |
| Анти-sp100, n (%) | 16/46 (34,8) | 4/14 (28,6) | 12/32 (37,5) | 0,739 |
| Анти-PML, n (%) | 5/46 (10,9) | 1/14 (7,1) | 4/32 (12,5) | 1,000 |
| Анти-SS-A, n (%) | 10/52 (19,2) | 3/18 (16,7) | 7/34 (20,6) | 1,000 |
| Ответ на лечение УДХК, n (%) | 49/91 (53,8) | 24/37 (64,9) | 25/54 (46,3) | 0,081 |
Печеночные неказеозные эпителиоидноклеточные гранулемы обнаруживали чаще у пациентов с ИПЛ (44,4% и 11,1%, р=0,016). У 7 из 8 пациентов с печеночными гранулемами выявили саркоидоподобное ИПЛ, у 1 – ОП. Цирроз печени реже формировался у пациентов с гранулемами в печени (27,3% и 61,8%, соответственно, р=0,081).
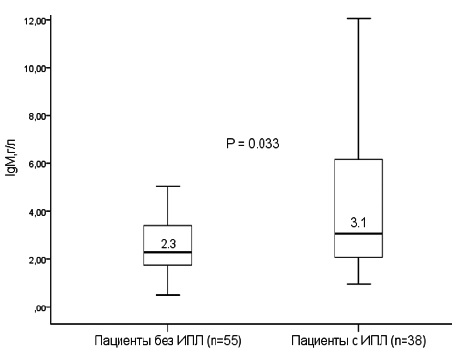
У больных с ИПЛ отмечали более высокие значения лейкоцитов крови, сывороточного уровня IgM (рис. 1) и чаще обнаруживали AMA-M2 в сыворотке крови, в то время как частота антител к gp210, sp100, SS-A и PML достоверно не отличалась между двумя группами. У AMA-M2-позитивных пациентов был выше титр сывороточного IgM (2,7 и 1,8 г/л, соответственно, р=0,05).
Многофакторный регрессионный анализ с коррекцией по полу, возрасту и статусу курения показал, что отсутствие симптомов заболевания печени в дебюте заболевания (отношение шансов [ОШ] 11,5; 95% доверительный интервал [ДИ] 1,2-109,4; p=0,033), наличие печеночных неказеозных эпителиоидноклеточных гранулем (ОШ 17,8; 95% ДИ 1,8-174,6; p=0,014), более высокие значения IgM сыворотки крови (ОШ 1,5; 95% ДИ 1,1-2,2; p=0,020) и лейкоцитов крови (ОШ 2,4; 95% ДИ 1,2-4,7; p=0,016) были независимыми факторами, ассоциировавшимися с наличием ИПЛ при ПБХ (табл. 2).
| Однофакторный анализ | Многофакторный анализ | |||
|---|---|---|---|---|
| ОШ (95% ДИ) | р | ОШ (95% ДИ) | p | |
| Возраст начала заболевания | 0,971 (0930-1,014) | 0,181 | - | - |
| Женский пол | 2,135 (0,214-21,336) | 0,519 | - | - |
| Ожирение | 3,055 (1,118-8,350) | 0,029 | - | - |
| Отсутствие печеночных симптомов в дебюте заболевания | 3,590 (1,474-8,743) | 0,005 | 11,509 (1,210-109,421) | 0,033 |
| Кожный зуд | 0,310 (0,126-0,765) | 0,011 | - | - |
| Гиперпигментация кожи | 0,239 (0,093-0,614) | 0,003 | - | - |
| Цирроз печени | 0,403 (0,173-0,941) | 0,036 | - | - |
| Гранулемы печени | 6,400 (1,402-29,209) | 0,017 | 17,754 (1,805-174,631) | 0,014 |
| Плотность печени | 0,852 (0,738-0,983) | 0,028 | - | - |
| Лейкоциты | 1,460 (1,134-1,881) | 0,003 | 2,356 (1,170-4,747) | 0,016 |
| Тромбоциты | 1,004 (0,999-1,009) | 0,088 | - | - |
| IgM | 1,254 (1,035-1,520) | 0,021 | 1,535 (1,067-2,208) | 0,020 |
| Серопозитивность по AMA-M2 | 1,825 (1,504-2,218) | 0,009 | - | - |
| Ответ на лечение УДХК | 2,142 (0,905-5,067) | 0,083 | - | - |
Течение интерстициального поражения легких. Дина мику рентгенологической картины оценивали у 34 пациентов с ИПЛ с интервалом в 3-6 мес. Шестнадцать пациентов получали комбинированную иммуносупрессивную терапию с применением системных глюкокортикостероидов в сочетании с азатиоприном или метотрексатом. Регресс ИПЛ наблюдали у 22 пациентов. При этом у 8 больных он был спонтанным (саркоидоподобный паттерн у 3, "субклиническое" ИПЛ у 3 и ОП у 2) и у 14 был индуцирован иммуносупрессивной терапией (саркоидоподобный паттерн у 8, OП у 4, неклассифицируемое ИПЛ у 2). Отсутствие изменений рентгенологической картины было зафиксировано у 2 пациентов, получавших иммуносупрессивную терапию (с саркоидоподобным паттерном и ОИП), и у 5 больных, не получавших иммуносупрессивные препараты ("субклиническое" ИПЛ у 4 и саркоидоподобный паттерн у 1). Рентгенологическое прогрессирование ИПЛ было констатировано у 5 пациентов. Двое из них (с НСИП и неклассифицируемым ИПЛ) умерли до начала лечения легочного поражения вследствие осложнений цирроза печени. У 2 пациентов (с ОП и неклассифицируемым ИПЛ) с незначительной отрицательной КТ-динамикой отсутствовали какие-либо респираторные симптомы и клиническое ухудшение, поэтому от назначения иммуносупреессивной терапии было решено воздержаться.
Исходы. Одна пациентка с КТ-паттерном НСИП имела прогрессирующее течение ИПЛ несмотря на проводимую иммуносупрессивную терапию и умерла от нарастающей дыхательной недостаточности. При аутопсии были выявлены признаки выраженного интерстициального фиброза легких, соответствующего гистологической картине фиброзирующего варианта НСИП. Таким образом, к моменту окончания исследования был зафиксирован только 1 неблагоприятный легочный исход.
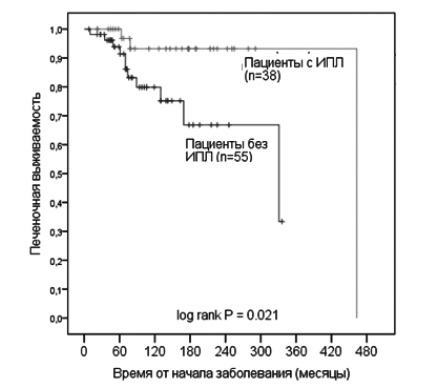
Во время исследования 12 пациентов умерли от осложнений цирроза печени, а 2 больным была проведена ортотопическая трансплантация печени. Трое умерших пациентов имели ИПЛ. Десятилетняя печеночная выживаемость у больных с ИПЛ была выше, чем у пациентов без ИПЛ (93,2% и 79,9%, соответственно, р=0,021, рис. 2). При однофакторном регрессионном анализе Кокса наличие ИПЛ было ассоциировано с меньшим риском наступления неблагоприятного печеночного исхода (отношение рисков 0,20; 95% ДИ 0,04-0,91; р=0,038), однако данный фактор утратил прогностическое значение при многофакторном регрессионном анализе (табл. 3).
| Однофакторный анализ | Многофакторный анализ | |||
|---|---|---|---|---|
| ОШ (95% ДИ) | р | ОШ (95% ДИ) | p | |
| Возраст начала заболевания | 1,038 (0,976-1,105) | 0,237 | 1,276 (1,078-1,510) | 0,005 |
| Цирроз печени | 57,644 (0,648-5128,411) | 0,077 | - | - |
| Асцит | 15,047 (4,087-55,399) | <0,001 | - | - |
| Варикозное расширение вен пищевода | 9,010 (1,985-40,893) | 0,004 | - | - |
| Печеночная энцефалопатия | 25,885 (7,042-95,147) | <0,001 | 53,950 (2,404-1210,699) | 0,012 |
| Альбумин | 0,834 (0,770-0,904) | <0,001 | - | - |
| Общий билирубин | 1,012 (1,007-1,016) | <0,001 | - | - |
| Протромбиновый индекс | 0,956 (0,931-0,983) | 0,001 | - | - |
| ИПЛ | 0,200 (0,044-0,911) | 0,038 | - | - |
| Ответ на лечение УДХК | 0,089 (0,018-0,437) | 0,003 | - | - |
Обсуждение
Результаты когортного исследования показали, что ИПЛ при ПБХ встречается гораздо чаще, чем считалось ранее, и может быть обнаружено почти у 41% пациентов с ПБХ при отсутствии сопутствующих СЗСТ. В ранее опубликованных работах частота ИПЛ у пациентов с ПБХ была ниже. В двух одноцентровых исследованиях, проведенных в Китае, различные варианты ИПЛ были диагностированы в 10,0% и 15,7% случаев, причем в оба исследования включали пациентов, имевших наряду с ПБХ и СЗСТ [18,25]. В первом исследовании факторами риска ИПЛ были синдром Рейно или СЗСТ [18], а во втором – ИПЛ ассоциировалось с наличием синдрома Шегрена [25]. В европейской популяции частота ИПЛ при ПБХ составляла от 2,2 до 5,4%, однако во всех исследованиях для скрининга ИПЛ использовали рентгенографию органов грудной клетки, что могло послужить причиной ложноотрицательного результата у части больных [26-28]. Одной из причин более высокой распространенности ИПЛ в обследованной нами когорте может быть наличие в выборке не только пациентов с изначально неизвестной легочной картиной, но и больных, уже имевших на момент включения в исследование диагностированное пульмонологом ПБХ-ассоциированное ИПЛ.
ИПЛ имело бессимптомное течение и было диагностировано только при КТ почти у трети пациентов. Симптомы поражения легких, обусловленные ИПЛ, были первым клиническим проявлением системной болезни почти у 10% пациентов, а ПБХ был заподозрен у них на основании наличия лабораторных признаков холестаза или специфических антител. Отсутствие печеночных симптомов в дебюте заболевания было независимым фактором риска ПБХ-ассоциированного ИПЛ, что позволяет относить больных ПБХ без клинических проявлений холестаза к группе повышенного риска ИПЛ. Исходя из того, что печеночное поражение при ПБХ-ассоциированном ИПЛ и, наоборот, ИПЛ при ПБХ могут иметь скрытое бессимптомное течение, следует вывод о необходимости исключения ПБХ у всех больных с впервые выявленным ИПЛ и проведения скрининга ИПЛ среди больных с ПБХ.
Как печеночные, так и внепеченочные гранулемы могут обнаруживаться у пациентов с ПБХ [29], особенно на ранних стадиях поражения печени [30,31]. В серии из 120 секционных наблюдений при ПБХ гранулемы в легких и внутригрудных лимфатических узлах были выявлены в 1,6% случаев [28]. Печеночные гранулемы при ПБХ в противоположность таковым при саркоидозе менее четко оформлены, не сливаются в конгломераты и локализуются преимущественно в пределах портального тракта вблизи пораженных междольковых желчевыводящих протоков [32]. Дуктопения, характерная для ПБХ, редко наблюдается при саркоидозе печени [32,33]. Как правило, пациенты с саркоидозом печени не имеют АМА-М2 и АНА, специфичных для ПБХ и демонстрируют уменьшение проявлений холестаза при лечении иммуносупрессивными препаратами, чего не наблюдается при ПБХ [34].
Патогенез и клиническое значение гранулематозного поражения печени при ПБХ остаются недостаточно изученными. R. Lee и соавт. показали более благоприятный прогноз течения поражения печени при ПБХ при наличии печеночных гранулем [35]. Z. You и соавт. предположили, что формирование печеночных гранулем при ПБХ может быть адаптивной реакцией в ответ на хроническое повреждение желчных протоков и позволяет уменьшить активность холангита [31]. Резуль таты нашего исследования показали, что цирроз печени формировался реже у пациентов с печеночными гранулемами, при этом у двух третей из них имелось саркодоподобное ИПЛ. Только 1 из 14 пациентов с данным типом ИПЛ умер от осложнений цирроза печени. Таким образом, нельзя исключать, что саркоидоподобная реакция при ПБХ может иметь протективный эффект в отношении поражения печени и способствует уменьшению скорости его прогрессирования.
У 9 (9,7%) из 93 пациентов мы выявили субклиническое ИПЛ, которое ранее было описано у пациентов с ревматоидным артритом [36], системной склеродермией, антисинтетазным синдромом и смешанным заболеванием соединительной ткани [37]. Предполагается, что этот КТ-паттерн представляет собой рентгенологическое проявление интерстициальных заболеваний легких на ранних этапах развития, в том числе ИПЛ, ассоциированного с СЗСТ [37-39]. Частота перехода субклинического ИПЛ в клинически явное при СЗСТ в разных исследованиях варьировалась от 34% до 57% при длительности наблюдения от 1,5 до 4,5 лет [37,40,41]. В нашем исследовании мы не зафиксировали ни одного случая прогрессирования субклинического ИПЛ до клинически значимого ИПЛ любого КТ-паттерна при медиане наблюдения 2,4 года. В 3 наблюдениях имело место обратное развитие субклинического ИПЛ при отсутствии какого-либо специфического лечения. Таким образом, скрининг ИПЛ в популяции пациентов с ПБХ позволяет не только выявить бессимптомно протекающее ИПЛ, но и диагностировать его на ранних этапах развития, когда легче контролировать течение заболевания и предотвратить его прогрессирование до терминальной стадии.
Как и в других исследованиях [25,42], наиболее час ты ми изменениями при гистологическом исследовании легочных биоптатов в нашей когорте были лимфоцитарная и эозинофильная инфильтрация альвеолярных перегородок и бронхиол, неказеозные эпителиодноклеточные гранулемы в лимфатических узлах, интерстиции и бронхиолах, фокусы ОП и интерстициальный легочный фиброз. Предполагается, что сочетание данных гистологических изменений, наблюдаемое у части пациентов с ПБХ-ассоциированным ИПЛ, может представлять собой особый гистологический паттерн поражения легких, специфичный для ПБХ [42].
Мы выявили статистически значимо более высокую частоту ожирения в группе пациентов с ИПЛ. В настоящее время установлена взаимосвязь между ожирением и некоторыми заболеваниями легких. Среди причин такой ассоциации рассматриваются легочные эффекты инсулинорезистентности, нарушения легочного микробиома и патологические иммунные процессы, опосредуемые такими медиаторами, как фактор некроза опухоли a, MCP-1 и трансформирующий фактор роста β1 [43-45]. Было показано, что больший объем висцеральной жировой ткани прямо коррелирует с более высокой распространенностью интерстициальных легочных изменений в популяции людей, страдающих ожирением [46]. Авторы предположили, что данная закономерность объясняется более высоким уровнем интерлейкина-6 и лептина у данных пациентов.
В нашем исследовании количество лейкоцитов и нейтрофилов периферической крови были выше у пациентов с ИПЛ, хотя их средние значения не выходили за пределы нормы. При многофакторном анализе более высокое количество лейкоцитов было независимым фактором, ассоциировавшимся с наличием ИПЛ. В нескольких исследованиях были продемонстрированы важная роль нейтрофилов в патогенезе интерстициальных заболеваний легких и их способность индуцировать развитие легочного интерстициального фиброза через различные механизмы [47-49]. Выше перечисленные факты и результаты нашего исследования позволяют предполагать участие нейтрофилов в патогенезе ПБХ-ассоциированного ИПЛ. С другой стороны, более высокое количество нейтрофилов и лейкоцитов в группе пациентов с ИПЛ может объясняться меньшей распространенностью цирротического гиперспленизма, если принять во внимание меньшую частоту цирроза печени и тенденцию к более высокому количеству тромбоцитов в группе пациентов с ИПЛ.
В нашем исследовании у всех пациенты с ИПЛ определялись AMA-M2, тогда как в группе пациентов без ИПЛ они отсутствовали в 16,4% случаев. М. Shen и соавт. также продемонстрировали более высокую распространенность АМА-М2 среди пациентов с ИПЛ (92,3% против 80,0%) [25]. В другой работе AMA-M2 были обнаружены у 10 (90,9%) из 11 пациентов с ПБХассоциированным ИПЛ [18]. Различия в частоте выявления АМА-М2 могут объясняться применением в нашем исследовании более чувствительной лабораторной методики на основе иммуноферментного анализа для детекции этого типа антител в сыворотке крови.
Как в настоящем исследовании, так и в работах других авторов было установлено, что пациенты с ПБХ, позитивные по АМА-М2, имеют более высокий уровень IgM сыворотки крови [50-52]. В настоящее время неизвестно, имеет ли IgM ту же антигенную специфичность, что и АМА-М2, у больных ПБХ. Было показано, что иммунные комплексы, циркулирующие в крови больных ПБХ, содержат АМА-М2 и антиген-мишень данных антител [53,54]. При этом по-прежнему окончательно не выяснено, является ли повышенный уровень IgM сыворотки крови при ПБХ лабораторным эпифеноменом или отражает участие IgM в формировании внепеченочных органных поражений при данном заболевании. В пользу последнего предположения свидетельствует то, что иммунные комплексы, содержащие IgM, были выявлены при гистологическом исследовании биоптатов кожи и почек пациентов, имеющих иммуноопосредованное поражение данных органов в рамках системных проявлений ПБХ [55-58]. Вопрос о способности IgM-содержащих иммунных комплексов индуцировать поражение легких при ПБХ остается открытым. Иммунные комплексы, содержащие IgG и C1q компонент комплемента, но не IgM, были выявлены в легочном интерстиции пациента с ПБХ-ассоциированным ИПЛ [59]. В нашем исследовании более высокий уровень IgM был независимым фактором риска развития ИПЛ, что позволяет предполагать вовлеченность IgM в процесс формирования легочного поражения при ПБХ. Таким образом, роль иммунокомплексных реакций в развитии ИПЛ при ПБХ в настоящее время остается не до конца раскрытой и должна стать предметом дальнейших исследований.
Роль АНА в развитии системных внепеченочных проявлений ПБХ изучена недостаточно. R. Nickowitz и соавт. не выявили корреляции между наличием антител к gp210 и ламину B и присутствием внепеченочных иммунных поражений за исключением ревматоидного артрита [60]. К сожалению, распространенность ИПЛ у пациентов с этими типами АНА не была представлена в данной публикации. М. Shen и соавт. не выявили отличий частоты АНА и антител к кардиолипину в группах пациентов с ИПЛ и без ИПЛ [25]. В настоящее время установлена роль антител к gp210 как независимого фактора риска неблагоприятного течения печеночного поражения при ПБХ [61]. В нашей работе не было выявлено статистически значимых различий частоты антител к gp210, sp100, PML и SS-A между группами пациентов, хотя и была отмечена тенденция к меньшей распространенности серопозитивности по анти-gp210 среди пациентов с ИПЛ. Мы предполагаем, что вышеперечисленные АНА направлены преимущественно против антигенов билиарного эпителия и не вовлечены в процесс формирования внепеченочных поражений.
Долгосрочный прогноз ИПЛ при ПБХ не изучался, хотя и были опубликованы наблюдения фатального течения ПБХ-ассоциированного ИПЛ [62,63]. В нашей когорте только 1 из 38 пациентов с ИПЛ умер от осложнений прогрессирующего фиброзирующего заболевания легких, диагностированного на продвинутой стадии, тогда как большинство пациентов имели стабильное течение легочного поражения и ответили на комбинированную иммуносупрессивную терапию системными глюкокортикостероидами в сочетании с азатиоприном или метотрексатом. Эти данные позволяют сделать вывод, что ПБХ-ассоциированное ИПЛ в большинстве случаев имеет благоприятный прогноз, если диагностировано и лечится на ранней стадии.
Десятилетняя выживаемость пациентов с ПБХ в различных исследованиях варьировалась от 63% до 94% [64]. В настоящее время неизвестно, является ли ИПЛ детерминантой прогноза печеночного поражения при ПБХ. М. Shen и соавт. не выявили какой-либо корреляции между наличием ИПЛ и риском в прогностической системе Мейо, которая используется для расчета выживаемости пациентов с ПБХ [25]. Мы не оценивали в настоящем исследовании риск неблагоприятного печеночного исхода по модели Мейо, однако десятилетняя выживаемость была выше у пациентов с ИПЛ, что было показано при однофакторном анализе выживаемости. Вместе с тем, при многофакторном анализе наличие ИПЛ не сохранило предиктивную значимость в отношении снижения риска смерти от осложнений цирроза печени или трансплантации печени, поэтому мы считаем, что ИПЛ при ПБХ не может считаться независимым фактором благоприятного печеночного прогноза.
Ограничениями нашего исследования были относительно небольшие объем выборки и продолжительность наблюдения, что могло повлиять на корректность анализа легочных и печеночных исходов. Количество легочных исходов было слишком малым для оценки легочной выживаемости и факторов риска неблагоприятных легочных исходов. Для валидации результатов нашего исследования необходимы проспективные многоцентровые исследования.
Заключение
Результаты нашего исследования продемонстрировали высокую распространенность ИПЛ, в том числе бессимптомного, среди пациентов с ПБХ и ПБХ-АИГ. В большинстве наблюдений ПБХ-ассоциированное ИПЛ имело доброкачественное течение и не определяло жизненный прогноз больного. Наличие ИПЛ было ассоциировано с меньшей тяжестью поражения печени. Биологические механизмы вовлечения легких при ПБХ и его влияние на печень как основной орган-мишень заболевания в настоящее время не раскрыты и должны стать предметом будущих исследований.
Используемые источники
- Kaplan M, Gershwin M. Primary biliary cirrhosis. N Engl J Med 2005;353(12): 1261–73.
- Lindor KD, Bowlus CL, Boyer J et al. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology (Baltimore, Md.) 2019;69(1):394–419.
- Rodriguez T, Latour E, Patel P, et al. Rare associations of primary biliary cirrhosis: a review article. Internet J Gastroenterol 2007;7(1).
- Floreani A, De Martin S, Secchi MF, Cazzagon N. Extrahepatic autoimmunity in autoimmune liver disease. Europ J Intern Med 2019;59:1–7.
- Wang L, Zhang F, Chen H, et al. Connective tissue diseases in primary biliary cirrhosis: a population-based cohort study. World J Gastroenterol 2013;19:5131–7.
- Strobel ES, Bonnet RB, Werner P, et al. Bronchiolitis obliterans organising pneumonia and primary biliary cirrhosis-like lung involvement in a patient with primary biliary cirrhosis. Clin Rheumatol 1998;17(3):246–9.
- Batista W, Simón-Talero R, García CA, et al. Cirrosis biliar primaria con neumonía organizada secundaria. Revista SOCAMPAR de Patología del Tórax 2017;2(3).
- Weissman E, Becker NH. Interstitial lung disease in primary biliary cirrhosis. Amer J Med Sci 1983;285(3):21–7.
- Koss M, Hochholzer L, Langloss J, et al. Lymphoid interstitial pneumonia: clinicopathological and immunopathological findings in 18 cases. Pathology 1987;19(2):178–85.
- Fujita J, Ohtsuki Y, Yoshinouchi T, et al. Idiopathic non-specific interstitial pneumonia: as an "autoimmune interstitial pneumonia". Respir Med 2005;99(2):234–40.
- Cavazza A, Rossi G, Corradi D, et al. Cellular non-specific interstitial pneumonia as a pulmonary manifestation of primary biliary cirrhosis. Pathology 2010;42:596–8.
- Iwata H, Atsuko I, Kadota J. Usual interstitial pneumonia associated with primary biliary cirrhosis. Respir Med 2008;1:216-8.
- Kishor S, Turner M, Borg B, et al. Cutaneous sarcoidosis and primary biliary cirrhosis: A chance association or related diseases? J Amer Acad Dermatol 2008;58(2):326–35.
- Wallaert B, Bonniere P, Prin L, et al. Primary biliary cirrhosis. Subclinical inflammatory alveolitis in patients with normal chest roentgenograms. Chest 1986;90(6):842–8.
- Spiteri MA, Johnson M, Epstein O, et al. Immunological features of lung lavage cells from patients with primary biliary cirrhosis may reflect those seen in pulmonary sarcoidosis. Gut 1990;31(2):208–12.
- Uddenfeldt P, Bjerle P, Danielsson A, et al. Lung function abnormalities in patients with primary biliary cirrhosis. Acta Med Scand 1988;223(6):549–55.
- Rodriguez-Roisin R, Pares A, Bruguera M, et al. Pulmonary involvement in primary biliary cirrhosis. Thorax 1981;36(3):208–12.
- Liu B, Zhang FC, Zhang ZL, et al. Interstitial lung disease and Sjogren's syndrome in primary biliary cirrhosis: a causal or casual association? Clin Rheumatol 2008;27(10):1299–306.
- Krowka MJ, Grambsch PM, Edell ES, et al. Primary biliary cirrhosis: relation between hepatic function and pulmonary function in patients who never smoked. Hepatology (Baltimore, Md.) 1991;13(6):1095–100.
- Costa C, Sambataro A, Baldi S, et al. Primary biliary cirrhosis: lung involvement. Liver 1996;15(4):196–201.
- Chazouillères O, Wendum D, Serfaty L, et al. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology (Baltimore, Md.) 1998;28(2):296–301.
- Corpechot C, ChazouillПres O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long-term outcome. J Hepatol 2011;55(6):1361–7.
- Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Europ Respir J 2005;26(2):319–38.
- Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Amer J Respir Crit Care Med 2013;188(6):733–48.
- Shen M, Zhang F, Zhang X. Primary biliary cirrhosis complicated with interstitial lung disease: a prospective study in 178 patients. J Clin Gastroenterol 2009;43:676–9.
- Lucey MR, Neuberger JM, Williams R. Primary biliary cirrhosis in men. Gut 1986;27(11):1373–6.
- Golding PL, Smith M, Williams R. Multisystem involvement in chronic liver disease. Studies on the incidence and pathogenesis. Amer J Med 1973;55(6):772–82.
- Stanley NN, Woodgate DJ. Mottled chest radiograph and gas transfer defect in chronic liver disease. Thorax 1972;27(3):315–23.
- Sherlock S, Fox RA, James DG, et al. Impaired delayed hypersensitivity in primary biliary cirrhosis. Lancet 1969;1:959–62.
- Drebber U, Mueller JJ, Klein E, et al. Liver biopsy in primary biliary cirrhosis: clinicopathological data and stage. Pathol Intern 2009;59(8):546–54.
- You Z, Wang Q, Bian Z, et al. The immunopathology of liver granulomas in primary biliary cirrhosis. J Autoimmu 2012;39(3):216–21.
- Lewis J. Histopathology of granulomatous liver disease. Clin Liv Dis 2018;11:77–80.
- Farouj NE, Cadranel JF, Mofredj A, et al. Ductopenia related liver sarcoidosis. World J Hepatol 2011;3(6):170–4.
- ?PF, Powers CR, Morris MJ. Pulmonary manifestations of primary autoimmune hepatobiliary disease. Clin Pulm Med 2005;12:232-45.
- Lee RG, Epstein O, Jauregui H, et al. Granulomas in primary biliary cirrhosis: a prognostic feature. Gastroenterology 1981;81(6):983–6.
- Doyle TJ, Dellaripa PF, Batra K, et al. Functional impact of a spectrum of interstitial lung abnormalities in rheumatoid arthritis. Chest 2014;146(1):41–50.
- Hoffmann-Vold AM, Andersson H, Reiseter S, et al. Subclinical interstitial lung disease is frequent and progresses across different connective tissue diseases. Ann Rheum Dis 2021;80:104-5.
- Yoo H, Hino T, Han J, et al. Connective tissue disease-related interstitial lung disease (CTD-ILD) and interstitial lung abnormality (ILA): Evolving concept of CT findings, pathology and management. Europ J Radiol 2020;8:100311.
- Panagopoulos P, Goules A, Hoffmann-Vold AM, et al. Natural history and screening of interstitial lung disease in systemic autoimmune rheumatic disorders. Ther Adv Musculoskelet Dis 2021;13:1759720X211037519.
- Gochuico BR, Avila NA, Chow CK, et al. Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch Intern Med 2008;168(2):159–66.
- Dawson JK, Fewins HE, Desmond J, et al. Predictors of progression of HRCT diagnosed fibrosing alveolitis in patients with rheumatoid arthritis. Ann Rheum Dis 2002;61(6):517–21.
- Lee HE, Churg A, Ryu JH, et al. Histopathologic findings in lung biopsies from patients with primary biliary cholangitis. Human Pathol 2018;82:177–86.
- Park YH, Oh EY, Han H, et al. Insulin resistance mediates high-fat diet-induced pulmonary fibrosis and airway hyperresponsiveness through the TGF-β1 pathway. Exp Mol Med 2019;51(5):1-12.
- Guo X, Sunil C, Qian G. Obesity and the development of lung fibrosis. Front Pharmacol 2022;12, 812166.
- Chioma OS, Hesse LE, Chapman A, Drake WP. Role of the microbiome in interstitial lung diseases. Front Med 2021;8:595522.
- Anderson MR, Kim JS, Allison M, et al. Adiposity and interstitial lung abnormalities in community-dwelling adults: The MESA Cohort Study. Chest 2021;160(2):582–94.
- Ding L, Yang J, Zhang C, et al. Neutrophils modulate fibrogenesis in chronic pulmonary diseases. Front Med 2021;8:616200.
- Zhang S, Jia X, Zhang Q, et al. Neutrophil extracellular traps activate lung fibroblast to induce polymyositis-related interstitial lung diseases via TLR9-miR-7-Smad2 pathway. J Cell Mol Med 2020;24(2):1658–69.
- Chrysanthopoulou A, Mitroulis I, Apostolidou E, et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol 2014;233:294–307.
- Lacerda MA, Ludwig J, Dickson ER, et al. Antimitochondrial antibody-negative primary biliary cirrhosis. Amer J Gastroenterol 1995;90(2):247–9.
- Sakauchi F, Mori M, Zeniya M, Toda G. Antimitochondrial antibody negative primary biliary cirrhosis in Japan: utilization of clinical data when patients applied to receive public financial aid. J Epidemiol 2006;16(1):30–4.
- Zhang FK, Jia JD, Wang BE. Clinical evaluation of serum antimitochondrial antibody-negative primary biliary cirrhosis. Hepatobil Pancr Dis Intern 2004;3(2):288–91.
- Gupta RC, McDuffie FC, Dickson ER, et al. The composition of the isolated soluble immune complexes in primary biliary cirrhosis (PBC). Gastroenterology 1978;75:967.
- Aibara N, Ohyama K, Nakamura, M, et al. Investigation of immune complexes formed by mitochondrial antigens containing a new lipoylated site in sera of primary biliary cholangitis patients. Clin Exp Immunol 2021;204(3):335–43.
- Lindgren S, Eriksson S, LЪfberg H, McKay J. IgM deposition in skin biopsies from patients with primary biliary cirrhosis. Acta Med Scand 1981;210(4):317–20.
- Rai GS, Hamlyn AN, Dahl MG, et al. Primary biliary cirrhosis, cutaneous capillaritis, and IgM-associated membranous glomerulonephritis. Brit Med J 1977;1(6064):817.
- Randle HW, Millns JL, Schroeter AL, Winkelmann RK. Cutaneous immunofluorescence in primary biliary cirrhosis. JAMA 1981;246(15):1679–81.
- Hendricks AA, Hutcheon DF, Maddrey WC, et al. Cutaneous immunoglobulin deposition in primary biliary cirrhosis. Arch Dermatol 1982;118(9):634–7.
- Wallace JG, Tong MJ, Ueki BH, Quismorio FP. Pulmonary involvement in primary biliary cirrhosis. J Clin Gastroenterol 1987;9(4):431–5.
- Nickowitz RE, Wozniak RW, Schaffner F, Worman HJ. Autoantibodies against integral membrane proteins of the nuclear envelope in patients with primary biliary cirrhosis. Gastroenterology 1994;106(1):193–9.
- Haldar D, Janmohamed A, Plant T, et al. Antibodies to gp210 and understanding risk in patients with primary biliary cholangitis. Liver Intern 2021;41(3):535–44.
- Hiraoka A, Kojima N, Yamauchi Y, et al. An autopsy case of primary biliary cirrhosis with severe interstitial pneumonia. Intern Med (Tokyo) 2001;40(11):1104-8.
- Osaka M, Aramaki T, Okumura H, Kawanami O. Primary biliary cirrhosis with fibrosing alveolitis. Gastroenterol Jap 1988;23(4):457–60.
- Lammers WJ, Kowdley KV, van Buuren HR. Predicting outcome in primary biliary cirrhosis. Ann Hepatol 2014;13(4):316–26.