Нефропатический цистиноз: механизмы развития и методы лечения
Нефропатический цистиноз – это редкое наследственное заболевание, связанное с накоплением кристаллов цистина в лизосомах клеток в результате мутации гена CTNS и дефицита транспортного белка цистинозина. Цистиноз характеризуется развитием дисфункции проксимальных канальцев (синдрома Фанкони) на первом году жизни и хронической почечной недостаточности, требующей заместительной почечной терапии, в детском или подростковом возрасте. В старшем возрасте у пациентов наблюдаются различные системные проявления, в том числе светобоязнь, гипотиреоз, сахарный диабет, гипогонадизм (у мужчин), миопатия с поражением мышц конечностей и дыхательных мышц, неврологические расстройства. У всех пациентов с цистинозом определяются отложения кристаллов цистина в роговице, которые могут быть выявлены при осмотре с помощью щелевой лампы. Для подтверждения диагноза измеряют концентрацию цистина в лейкоцитах и проводят молекулярно-генетическое исследование. Важность своевременной диагностики нефропатического цистиноза определяется возможностью патогенетической терапии цистеамином, который обеспечивает выведение цистина из лизосом и позволяет задержать прогрессирование хронической почечной недостаточности и поражения других органов.
Цистиноз – это редкая аутосомнорецессивная лизосомная болезнь накопления, которая является одной из основных причин синдрома Фанкони у детей [1-4]. Причина цистиноза – мутации гена CTNS, кодирующего белок цистинозин, который переносит аминокислоту цистин через мембрану лизосом [5]. Нарушение функции этого транспортного белка приводит к накоплению и кристаллизации цистина в лизосомах клеток и повреждению почек и других органов [6]. Основным клиническим вариантом цистиноза является инфантильная нефропатическая форма, которая сопровождается развитием хронической почечной недостаточности, требующей заместительной почечной терапии, в детском возрасте, хотя у небольшой части пациентов (менее 5%) тяжелое поражение почек отмечается в подростковом возрасте или отсутствует [4]. Как и другие лизосомные болезни накопления, цистиноз – это системное заболевание, характеризующееся поражением не только почек, но и других органов и тканей, в том числе глаз, желез внутренней секреции, мышц, нервной системы, поэтому нередко использующийся термин "нефропатический цистиноз" условный.
Цистиноз относится к орфанным заболеваниям – заболеваемость составляет 0,5-1 на 100000 новорожденных, хотя в некоторых популяциях, например, в Бретани (Франция) и Квебеке (Канада), она оказалась в несколько раз выше [5]. Нельзя исключить, что данные эпидемиологических исследований о распространенности цистиноза могут быть заниженными, так как они основываются на числе зарегистрированных случаев, а не результатах скрининга среди новорожденных. В США в 2010 г. доля пациентов с цистинозом составила 1,4% среди детей, получавших лечение диализом, и 2,1% среди детей, перенесших трансплантацию почки [7].
Симптомы нефропатического цистиноза появляются в первые годы жизни, однако это заболевания не следует считать "педиатрической" проблемой, учитывая значительное увеличение выживаемости пациентов благодаря заместительной почечной терапии и патогенетической терапии цистеамином, возможность поздней диагностики заболевания даже при наличии типичных симптомов и системность проявлений, которая обычно проявляется в подростковом или старшем возрасте.
Этиология и патогенез
К настоящему времени описаны более 140 мутаций гена CTNS, расположенного на хромосоме 17p13.2 [8]. В странах Европы и Северной Америки чаще всего встречается делеция 57257 нуклеотидов, которая наблюдается примерно у половины пациентов с цистинозом. В российской популяции та же мутация была выявлена у 25% из 40 детей с цистинозом [9]. Тяжесть течения цистиноза зависит от типа мутаций, в частности инфантильный нефропатический цистиноз обычно развивается при наличии делеций и других мутаций, сопровождающихся полным отсутствием функционального белка [10].
Непосредственной причиной проксимальной ка наль цевой дисфункции при цистинозе считают образование кристаллов цистина, которое начинается, когда его концентрация в лизосомах превышает пороговый уровень, составляющий около 5 ммоль. Однако результаты экспериментальных исследований свидетельствуют о том, что патогенез нефропатического цистиноза более сложный и предполагает участие не только прямого повреждения клеток проксимальных канальцев под действием кристаллов цистина, но и других механизмов [11]. Например, во многих клетках, в том числе эпителиальных клетках проксимальных канальцев, фибробластах и подоцитах, при цистинозе определялись усиление апоптоза [12], а также дефицит АТФ и цАМФ, сопровождавшийся дисфункцией митохондрий [13]. Недостаточность цистинозина вызывает нарушение сигнальной системы mTORC1 (mammalian target of rapamycin complex 1 – мишень рапамицинового комплекса 1 у млекопитающих) и подавление аутофагии [14]. Важное значение в развитии поражения проксимальных канальцев и других тканей при цистинозе придают воспалению [15]. У пациентов с цистинозом и Ctns-/- мышей кристаллы цистина были обнаружены в макрофагах в большинстве органов, включая почки, костный мозг, печень, кожу и желудочно-кишечный тракт. Накопление цистина, связанное с дефицитом цистинозина и фагоцитозом содержимого погибающих клеток, вызывает активацию моноцитов/макрофагов, которые выделяют провоспалительные цитокины, такие как интерлейкин-1β [16]. Полагают, что образование кристаллов, в том числе цистина, может индуцировать клеточный некроптоз, сопровождающийся выделением молекулярных фрагментов, ассоциированных с повреждением (DAMP) [17]. Последние инициируют воспалительный ответ путем активации паттерн-распознающих рецепторов (PRR), в том числе Toll- и Nod-подобных рецепторов, которые экспрессируются в клетках врожденной иммунной системы, и образования инфламмасомы – макромолекулярного комплекса, состоящего из белка NLRP3, вспомогательного белка ASC и прокаспазы-1. Последняя превращается в активную каспазу-1 и вызывает активацию и секрецию провоспалительных цитокинов ИЛ-1β и ИЛ-18 [18]. Кроме того, NLRP3 активирует трансформирующий фактор роста-β, что способствует развитию фиброза [19], причем этот эффект не зависит от секреции провоспалительных цитокинов.
Клиническая картина
Ведущим в клинической картине нефропатического цистиноза является поражение почек, которое приводит к развитию хронической почечной недостаточности, требующей заместительной почечной терапии, в детском возрасте (рис. 1). В подростковом или старшем возрасте развивается и постепенно прогрессирует поражение других органов, хотя необходимо учитывать, что некоторые системные проявления могут быть следствием не только цистиноза, но и хронической почечной недостаточности [20].
![Клинические проявления нефропатического цистиноза у детей, подростков и взрослых [20]](/wp-content/uploads/2023/04/nefropaticheskij-cistinoz-mekhanizmy-razvitiya-i-metody-lecheniya_fig1.jpg)
Почки. Инфантильный нефропатический цистиноз проявляется дисфункцией проксимальных почечных канальцев (синдром Фанкони), которая отмечается на первом году жизни и приводит к увеличению экскреции с мочой аминокислот, глюкозы, фосфора, бикарбоната, натрия, калия, кальция, мочевой кислоты и карнитина [21]. У детей цистиноз является одной из основных причин синдрома Фанкона, в то время как у взрослых проксимальная канальцевая дисфункция обычно развивается под действием лекарственных средств и тяжелых металлов или при заболеваниях, сопровождающихся диспротеинемией (множественная миелома, синдром Шегрена, амилоидоз). Клинические проявления дисфункции проксимальных почечных канальцев включают в себя жажду, полидипсию и полиурию, эпизоды дегидратации, задержку роста, мышечную слабость и рахит, а лабораторные – аминоацидурию, глюкозурию (при отсутствии гипергликемии, что имеет диагностическое значение), гипокалиемию, метаболический ацидоз, гипофосфатемию, гипоурикемию и реже гипонатриемию. Помимо задержки роста, характерное проявление синдрома Фанкони у детей – гипофосфатемический рахит, который обусловлен потерей фосфора и витамин D связывающего белка с мочой и нарушением конверсии витамина D на фоне снижения активности β1-гидроксилазы в проксимальных почечных канальцах. Протеинурия обычно минимальная. Со временем отмечается постепенное снижение скорости клубочковой фильтрации (СКФ), которое приводит к формированию терминальной стадии хронической болезни почек в возрасте 8-12 лет. По данным ретроспективного исследования у 205 больных цистинозом, обследованных до появления патогенетической терапии, медиана возраста пациентов на момент смерти от уремии или инициации заместительной почечной терапии составила 9,2 года [22]. При ювенильном цистинозе функция почек ухудшается медленнее, а терминальная хроническая почечная недостаточность развивается в старшем возрасте (от 12 до 28 лет) [23].
Орган зрения. Первое внепочечное проявление цистиноза – отложение кристаллов цистина в роговице, которое определяется при осмотре с помощью щелевой лампы к возрасту 16 мес у всех пациентов и является важным диагностическим признаком. Первоначально отложение цистина не сопровождается симптомами, однако в возрасте 8-12 лет появляется фотофобия. Цистин откладывается и в других отделах глазного яблока, в том числе сетчатке, что приводит к слепоте у 10-15% больных [24]. У взрослых в редких случаях фотофобия может быть единственным проявлением цистиноза [23].
Эндокринная система. Накопление цистина в лизосомах клеток различных тканей, в том числе желез внутренней секреции, начинается в детском возрасте и постепенно приводит к их атрофии и фиброзу и нарушению функции органов. Чаще всего наблюдается поражение щитовидной (17-89%) и поджелудочной (754%) желез, сопровождающееся развитием первичного гипотиреоза и сахарного диабета, соответственно [25]. Вариабельность частоты поражения этих органов в когортных исследованиях отражает различия медианы возраста обследованных пациентов и доли больных, получавших патогенетическую терапию цистеамином, которая задерживает прогрессирование заболевания. У мальчиков обычно наблюдается первичный гипогонадизм, в то время как у женщин репродуктивная функция, как правило, сохраняется. A. Servais и соавт. описали 19 случаев беременности у 12 пациенток с нефропатическим цистинозом (в большинстве случаев после трансплантации почки) [26]. В 13 (68,4%) случаях беременность завершилась рождением живых детей. Практически у всех пациентов с инфантильным нефропатическим цистинозом определяется низкий рост, который обычно на 2-3 стандартных отклонений ниже среднего для соответствующего возраста. Нарушение линейного роста может быть следствием не только самого заболевания, но и хронической почечной недостаточности, так как улучшение роста отмечалось как на фоне терапии цистеамином, так и после трансплантации почки [25].
W. Gahl и соавт. проанализировали частоту внепочечных проявлений у 100 взрослых пациентов с цистинозом, возраст которых варьировался от 18 до 45 лет [27]. Большинство из них перенесли трансплантацию почки (92%). В этом исследовании частота гипотиреоза составила 92%, гипогонадизма у мужчин – 75%, инсулинзависимого сахарного диабета – 33%. Треть больных умерли в возрасте в среднем 28,5 лет.
Желудочно-кишечный тракт. По данным систематизированного обзора клинических исследований [25], у большинства пациентов с цистинозом наблюдаются тошнота и рвота, которые обычно возникают эпизодически и иногда со временем проходят. Многие больные жалуются на снижение аппетита, запор, диарею и боли в животе. Характерны нарушения глотания и гастроэзофагеальный рефлюкс, которые возникают в результате поражения мышечной ткани. Возможны также увеличение печени и селезенки и развитие портальной гипертензии, не связанной с формированием цирроза печени [28]. При биопсии печени у пациентов с цистинозом обнаруживали отложение цистина в купфферовских клетках, которое может быть причиной нодулярной гиперплазии этого органа.
Нервная система. Тяжелые неврологические нарушения нечасто встречаются при цистинозе, хотя в когортных исследованиях частота поражения центральной нервной системы (ЦНС) составляла от 3 до 27% [25]. Периферическая нервная система при цистинозе не поражается. Проявления энцефалопатии при этом заболевании включают в себя увеличение внутричерепного давления, мозжечковые и пирамидные признаки, когнитивные расстройства и псевдобульбарный паралич [29,30]. Могут наблюдаться и проявления инсульта, такие как гемиплегия и кома. Развитие поражения ЦНС при цистинозе связывают с отложением цистина, которое приводит к развитию кальциноза и атрофии ткани головного мозга. Следует учитывать, что у пациентов, перенесших трансплантацию почки, длительная иммуносупрессивнная терапия может осложниться оппортунистическими инфекциями, такими как криптококковый менингит.
Миопатия. Частота поражения мышц при цистинозе у взрослых составляет 24-69% [25]. Миопатия в первую очередь проявляется слабостью в дистальных мышцах верхних конечностей, хотя со временем возможно вовлечение и проксимальных мышц, а также мышц нижних конечностей. Активность КФК нормальная или повышена. У 2/3 взрослых пациентов с цистинозом развивается рестриктивная дыхательная недостаточность при отсутствии рентгенологических изменений в легких [27]. Выраженность нарушения функции внешнего дыхания коррелировала с тяжестью миопатии [31], поэтому основной причиной дыхательной недостаточности при цистинозе считают поражение дыхательных мышц, включая диафрагму, хотя определенное значение могут иметь и изменения грудной клетки, связанные с рахитом и задержкой линейного роста. Миопатия сопровождается также нарушением глотания, которое наблюдается у большинства пациентов старшего возраста.
Сердечно-сосудистая система. У взрослых пациентов с цистинозом нередко определяется кальциноз артерий, который может быть причиной сердечно-сосудистых осложнений, развивающихся в молодом возрасте [1]. M. Ueda и соавт. выявили кальциноз артерий, преимущественно коронарных, с помощью компьютерной томографии у 32% из 41 пациента с цистинозом, перенесшего трансплантацию почки [32]. Паценты с кальцинозом артерий были старше и чаще страдали сахарным диабетом, однако длительность лечения диализом не отличалась между группами больных, у которых определялись или отсутствовали отложения кальция в стенках артерий, что может указывать на роль отложения цистина в патогенезе кальциноза артерий. Причиной сердечно-сосудистых осложнений при нефропатическом цистинозе может быть и хроническая болезнь почек, учитывая многолетний анамнез хронической почечной недостаточности у пациентов даже молодого возраста. Z. Modi и соавт. изучили сердечнососудистую заболеваемость и смертность более чем у 33000 молодых пациентов с различными заболеваниями почек, начавших заместительную почечную терапию в возрасте от 1 до 30 лет [33]. Доля сердечно-сосудистых причин в структуре общей смертности составила 37,7% и была выше всего в группе пациентов в возрасте 21-29 лет (39,1%). Риск сердечно-сосудистой смерти увеличивался с возрастом и у молодых людей был достоверно выше, чем у подростков и детей. Другими факторами риска смерти от сердечно-сосудистых заболеваний были низкий индекс массы тела и различные сопутствующие заболевания, в частности сахарный диабет, в то время как трансплантация почки ассоциировалась с улучшением сердечно-сосудистых исходов по сравнению с лечением диализом.
Диагноз
Предполагать нефропатический цистиноз следует у всех больных с хронической почечной недостаточностью, развившейся в детском или подростковом возрасте, особенно при наличии анамнестических указаний на синдром Фанкони в первые годы жизни. Как указано выше, большинство пациентов с нефропатическим цистинозом начинают заместительную почечную терапию в возрасте до 18 лет. Простым скрининговым методом является осмотр роговицы с помощью щелевой лампы, который позволяет выявить отложения цистина. Последние определяются у всех пациентов с цистинозом (старше 1,5 лет) и отсутствуют при других наследственных заболеваниях, вызывающих синдром Фанкони у детей (тирозинемия, галактоземия, гепаторенальный гликогеноз, болезнь Дента, синдром Лове и др.) [2]. Отложения цистина могут быть выявлены в биоптате почки или других тканей, однако биопсию не считают необходимой для диагностики нефропатического цистиноза. Для подтверждения диагноза определяют содержание цистина в лейкоцитах крови с помощью высокоэффективной жидкостной хроматографии или жидкостной хроматографии/тандемной массспектрометрии и проводят молекулярно-генетическое исследование, позволяющее выявить мутации гена CTNS [34].
Быстрое развитие тяжелого и необратимого поражения почек и возможность эффективной патогенетической терапии оправдывают проведение скрининга цистиноза у новорожденных, хотя при обсуждении таких программ приходится учитывать экономические аспекты их внедрения, особенно если речь идет об очень редких заболеваниях. В Германии был проведен скрининг у 257734 новорожденных на основании анализа трех мутаций гена CTNS, которые чаще всего встречаются у немцев (75% случаев цистиноза) [35]. Цистиноз был диагностирован у одного ребенка, которому был назначен цистеамин на 18-й день после рождения. Через 16 мес признаки синдрома Фанкони у ребенка отсутствовали.
При наличии в семье одного ребенка с цистинозом при каждой следующей беременности рекомендуется пренатальная диагностика болезни с помощью измерения уровня свободного цистина в культуре амниоцитов или клетках ворсин хориона или генетического исследования.
Лечение
Цистеамина битартрат. Основа лечения нефропатического цистиноза – применение цистеамина битартрата,который используется при этом заболевании с 80-х гг.прошлого столетия. Цистеамин проникает в лизосомы,в которых он расщепляет цистин на две молекулыцистеина и соединяется с одной из них с помощью дисульфидного мостика. Цистеин и комплекс цистеинаи циастеамина транспортируются через мембрану лизо-сомы белком PQLC2, т.е. они не нуждаются в цистино-зине для выхода из лизосом. Таким образом, лечениецистеамином вызывает выведение цистина из лизосомклеток и предупреждает дальнейшее его накопление.Препарат выпускается в виде капсул для приема внутрьи глазных капель, содержащих 0,55% раствор цистеами-на гидрохлорида. Капли применяют для растворениякристаллов цистина в роговице, так как пероральнаятерапия цистеамином не оказывает на них влияние. Вклинических исследованиях регулярное введениекапель цистеамина вызывало уменьшение фотобоязнии поддерживало нормальную остроту зрения [36,37].
Пероральная терапия цистеамином позволяет задержать развитие терминальной хронической почечной недостаточности на 6-10 лет и предупреждает или тормозит развитие внепочечных проявлений цистиноза [6]. В ретроспективном исследовании эффективность длительной терапии цистеамином изучали у 86 взрослых больных нефропатическим цистинозом (средний возраст 26,7 лет) [38]. У 75 из них лечение продолжали в течение в среднем 17,4 лет. Терапия цистеамином, начатая в возрасте до 5 лет, значительно задерживала развитие терминальной хронической почечной недостаточности (на 6-7 лет), а также гипотиреоза, сахарного диабета и нейромышечных нарушений. У пациентов, начавших лечение после 5 лет, сроки развития сахарного диабета и гипотиреоза, значительно увеличились по сравнению с таковыми у нелеченных пациентов. Кроме того, терапия цистеамином привела к увеличению продолжительности жизни больных цистинозом. В другом когортном исследовании у больных, продолжавших терапию цистеамином более 20 лет, частота сахарного диабета и миопатии снизилась с 28% до 0% и с 60% до 0%, соответственно, а у больных, получавших препарат в течение более 8 лет, частота гипотиреоза снизилась с 87% до 56% [27].
Хотя пероральная терапия цистеамином обоснована в любые сроки после установления диагноза нефропатического цистиноза, в том числе у взрослых пациентов, у которых она по крайней мере позволяет избежать дальнейшего ухудшения состояния, тем не менее, начинать лечение лучше как можно раньше, когда отсутствует необратимое повреждение органов и тканей. K. Hohenfellner и соавт. показали, что терапия цистеамином, начатая в первые 2 мес жизни у 4 пациентов с инфантильным нефропатическим цистинозом, вызывала значительное уменьшение проявлений синдрома Фанкони [39]. Эти данные не подтверждают общепринятое мнение о том, что патогенетическая терапия не влияет на состояние проксимальных почечных канальцев, которые при цистинозе быстро поражаются уже в первые месяцы после рождения, хотя на практике ранняя диагностика заболевания до развития развернутых проявлений проксимальной канальцевой дисфункции возможна только путем скрининга среди новорожденных.
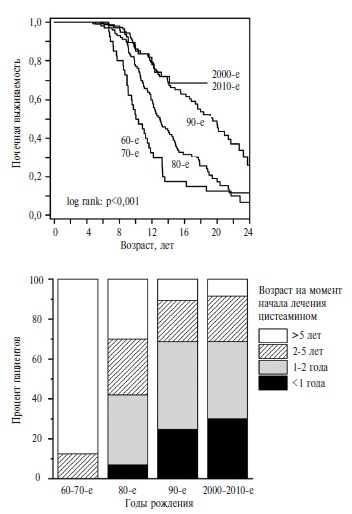
F. Emma и соавт. в международном исследовании, проведенном в странах Европы и Турции, изучили исходы нефропатического цистиноза у 453 пациентов, родившихся в 1964-2016 гг. [40]. Медиана длительности наблюдения составила 15,3 года. У 89% больных лечение цистеамином было назначено (медиана возраста – 1,6 года) до начала заместительной почечной терапии. В качестве "исторического" контроля использовали результаты исследования, в котором оценивали динамику сывороточного уровня креатинина у пациентов с нефропатическим цистинозом до появления цистеамина в Европе [41]. За последние десятилетия авторы выявили значительное увеличение доли пациентов, начавших лечение цистеамином в течение первых 1-2 лет жизни (рис. 2). Параллельно возросла почечная выживаемость пациентов с нефропатическим цистинозом (p<0,001). Медиана прироста ее составила 9,1 года. По данным многофакторного регрессионного анализа, только возраст, в котором пациенты начинали лечение цистеамином, и средние уровни цистина в лейкоцитах ассоциировались с более поздним развитием хронической болезни почек 5 стадии. Последний факт подтверждает важность мониторирования эффективности терапии цистеамином путем определения концентрации цистина в лейкоцитах, которая не должна превышать 1 нмоль 1/2 цистина на мг белка [27].
Заместительная почечная терапия. Метод выборазаместительной почечной терапии у детей – трансплан тация почки. C. Cohen и соавт. сравнили отдаленныеисходы трансплантации почки у 30 взрослых пациентовс цистинозом и 93 больных контрольной группы, подо бранных по возрасту и ряду других показателей [42]. Восновной группе трансплантация была выполнена ввозрасте от 7 до 36,5 лет (медиана – 20,4 года).Выживаемость трансплантата у больных цистинозомбыла выше, чем в контрольной группе (р=0,013), а мно гофакторный анализ подтвердил, что наличие этогозаболевания ассоциируется со снижением риска егоотторжения (отношение рисков 0,11; 95% доверитель ный интервал 0,02-0,61). Частота посттрансплантацион ного сахарного диабета в основной группе была выше,чем в контрольной (13,0% и 5,0%), однако различия недостигли статистической значимости. Следует подчерк нуть, что синдром Фанкони в трансплантате не рециди вирует [2].
Блокаторы ренин-ангиотензиновой системы. Инги биторы АПФ и блокаторы ангиотензиновых рецепторов широко используются для замедления темпа снижения скорости клубочковой фильтрации у пациентов с заболеваниями почек, особенно сопровождающимися протеинурией. Высказано предположение о том, что они могут применяться и у детей с нефропатическим цистинозом [43]. Однако эффективность блокаторов ренинангиотензиновой системы у таких больных не доказана. Более того, они могут ухудшить перфузию почек, которая обычно снижена у больных с синдромом Фанкони. В связи с этим применять подобные препараты у пациентов с цистинозом следует с осторожностью.
Индометацин. В международном европейском исследовании у 43% пациентов с нефропатическим цистинозом для уменьшения проявлений синдрома Фанкони применяли индометацин, который уменьшает полиурию и усиливает реабсорцию соли в петле Генле и собирательных канальцах [40]. В этом исследовании не было выявлено ухудшения почечных исходов на фоне длительного приема индометацина, хотя он может вызвать снижение СКФ у пациентов с нарушенной перфузией почек, так как увеличение продукции простагландинов является компенсаторным механизмом. Кроме того, индометацин может оказывать токсическое действие на интерстиций почек и вызывает желудочнокишечные нарушения. По мнению F. Emma и соавт. [40], индометацин целесообразно применять только в первые годы жизни, когда у пациентов с нефропатическим цистинозом отмечаются выраженная полиурия и потеря электролитов с мочой. Препарат следует отменить в случае развития дегидратации, артериальной гипотонии или ухудшения функции почек [44].
Заключение
Нефропатический цистиноз – это редкое наследственное заболевание, которое следует исключать у всех детей с синдромом Фанкони, а также у больных с терминальной уремией, развивающейся в детском или подростковом возрасте. Помимо поражения почек и органа зрения, проявляющегося светобоязнью, у взрослых пациентов наблюдаются различные системные проявления, в том числе гипотиреоз, сахарный диабет, поражение мышц конечностей и дыхательных мышц и др. У всех пациентов с цистинозом определяются отложения цистина в роговице, которые могут быть выявлены с помощью щелевой лампы. Для подтверждения диагноза измеряют концентрацию цистина в лейкоцитах и проводят молекулярно-генетическое исследование с целью выявления мутации гена CTNS. Всем больным цистинозом показана пожизненная терапия цистеамином для профилактики развития или прогрессирования инвалидизирующих проявлений заболевания.
Используемые источники
- Чеботарева Н.В., Цыгин А.Н., Буланов Н.М. и др. Цистиноз: патогенез, клинические проявления и лечение. Клин фармакол тер 2021;30(1):80-88. [Chebotareva N, Tsygin A, Bulanov N, et al. Cystinosis: pathogenesis, clinical features and treatment. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(1):80-88 (In Russ.)].
- Цыгин А.Н., Каган М.Ю., Картамышева Н.Н. и др. Нефропатический цистиноз. Недооцененная проблема детской нефрологии. Клиническая нефрология 2011;4:20-3 [Tsygin AN, Kagan Myu, Kartamysheva NN, et al. Nephropathic cystinosis. An underestimated problem in pediatric nephrology. Clinical Nephrology 2011;4:20-3 (In Russ.)].
- Чеботарева Н.В., Цыгин А.Н., Буланов Н.М. и др. Синдром Фанкони у взрослых и детей. Клин фармакол тер 2022;31(1): 69-74 [Chebotareva N, Tsygin A, Bulanov N, et al. Fanconi syndrome in adults and children. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(1):69-74 (In Russ.)].
- Langman CB, Barshop BA, DeschРnes G, et al. Controversies and research agenda in nephropathic cystinosis: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int 2016;89(6): 1192-203.
- Jamalpoor A, Othman A, Levtchenko EN, et al. Molecular mechanisms and treatment options of nephropathic cystinosis. Trends Mol Med 2021;27(7):673-86.
- Elmonem MA, Veys KR, Soliman NA, et al. Cystinosis: a review. Orphanet J Rare Dis 2016;11:47.
- North American Pediatric Renal Trials and Collaborative Studies. NAPRTCS Annual Reports. NAPRTCS Online [online]. 2011. https://web.emmes.com/study/ped/annlrept/annlrept.html.
- David D, Princiero Berlingerio S, et al. Molecular basis of cystinosis: geographic distribution, functional consequences of mutations in the CTNS gene, and potential for repair. Nephron 2019;141(2):133-46.
- Savostyanov KV, Pushkov AA, Shchagina OA, et al. Genetic landscape of nephropathic cystinosis in Russian children. Front Genet 2022;13:863157.
- Attard M, Jean G, Forestier L, et al. Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin. Hum Mol Genet 1999;8:2507–14.
- Cherqui S, Courtoy PJ. The renal Fanconi syndrome in cystinosis: pathogenic insights and therapeutic perspectives. Nat Rev Nephrol 2017;13(2):115–31.
- Park MA, Pejovic V, Kerisit KG, et al. Increased apoptosis in cystinotic fibroblasts and renal proximal tubule epithelial cells results from cysteinylation of protein kinase Cdelta. J Am Soc Nephrol 2006;17(11):3167-75.
- Bellomo F, Signorile A, Tamma G, et al. Impact of atypical mitochondrial cyclicAMP level in nephropathic cystinosis. Cell Mol Life Sci 2018;75(18):3411-22.
- Ivanova EA, van den Heuvel LP, Elmonem MA, et al. Altered mTOR signalling in nephropathic cystinosis. J Inherit Metab Dis 2016;39(3):457-64.
- Elmonem MA, Veys KRP, Prencipe G. Nephropathic cystinosis: pathogenic roles of inflammation and potential for new therapies. Cells 2022;11(2):190.
- Prencipe G, Caiello I, Cherqui S, et al. Inflammasome activation by cystine crystals: implications for the pathogenesis of cystinosis. J Am Soc Nephrol 2014;25:1163-9.
- Mulay SR, Desai J, Kumar SV, et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat Commun 2016;7:10274.
- Mulay SR, Shi C, Ma X, Anders HJ. Novel insights into crystal-induced kidney injury. Kidney Dis 2018;4:49–57.
- Wang W, Wang X, Chun J, et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J Immunol 2013;190:1239–49.
- Levtchenko E, Servais A, Hulton SA, et al. Expert guidance on the multidisciplinary management of cystinosis in adolescent and adult patients. Clin Kidney J 2022;15(9):1675–84
- Foreman J. Fanconi syndrome. Pediatr Clin N Am 2019;66:159–167
- Gretz N, Manz F, Augustin R, et al. Survival time in cystinosis. A collaborative study. Proc Eur Dial Transplant Assoc 1983;19:582-9.
- Servais A, Morinière V, Grunfeld JP, et al. Late-onset nephropathic cystinosis: clinical presentation, outcome, and genotyping. Clin J Am Soc Nephrol 2008;3:27–35.
- Gahl WA, Kuehl EM, Iwata F, et al. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eye drops. Mol Genet Metab 2000;71:100–20.
- Kasimer RN, Langman CB. Adult complications of nephropathic cystinosis: a systematic review. Pediatr Nephrol 2021;36(2):223-36.
- Servais A, Janssen MCH, Blakey H, et al; Metabolic Nephropathy Workgroup of the European Reference Network for Rare Kidney Diseases (ERKNet) and the ERA working groups on inherited kidney diseases (WGIKD). Pregnancy in cystinosis patients with chronic kidney disease: A European case series. J Inherit Metab Dis 2022;45(5):963-8.
- Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med 2007;147(4):242-50.
- DiDomenico P, Berry G, Bass D, et al. Noncirrhotic portal hypertension in association with juvenile nephropathic cystinosis: case presentation and review of the literature. J Inherit Metab Dis 2004;27(5):693-9.
- Broyer M, Tête MJ, Guest G, et al. Clinical polymorphism of cystinosis encephalopathy. Results of treatment with cysteamine. J Inherit Metab Dis 1996;19:65–75.
- Vogel DG,Malekzadeh MH, Cornford ME, et al. Central nervous system involvement in nephropathic cystinosis. J Neuropathol Exp Neurol 1990;49:591–599
- Anikster Y, Lacbawan F, Brantly M, et al. Pulmonary dysfunction in adults with nephropathic cystinosis. Chest 2001;119:394–401.
- Ueda M, O’Brien K, Rosing DR, et al. Coronary artery and other vascular calcifications in patients with cystinosis after kidney transplantation. Clin J Am Soc Nephrol 2006;1:555–62.
- Modi ZJ, Lu Y, Ji N, et al. Risk of cardiovascular disease and mortality in young adults with end-stage renal disease: An analysis of the US Renal Data System. JAMA Cardiol 2019;4(4):353-62.
- Савостьянов К.В., Мазанова Н.Н., Пушков А.А. и др. Хромато-масс-спектрометрическая и молекулярно-генетическая диагностика цистиноза у российских детей. Педиатрия 2018;97(5):71-8 [Savostyanov KV, Mazanova NN, Pushkov AA, et al. Chromatography-mass spectometry and molecular genetic diagnosis of cystinosis in Russian children. Pediatriya 2018;97(5):71-8 (In Russ.)].
- Hohenfellner K, Bergmann C, Fleige T, et al. Molecular based newborn screening in Germany: Follow-up for cystinosis. Mol Genet Metab Rep 2019;21:100514.
- Kaur S, Sarma P, Kaur H, et al. Efficacy and safety of topical cysteamine in corneal cystinosis: a systematic review and meta-analysis. Am J Ophthalmol 2020;S0002-9394(20)30418-9.
- Liang H, Labbé A, Baudouin C, et al. Long-term follow-up of cystinosis patients treated with 0.55% cysteamine hydrochloride. Br J Ophthalmol 2020;bjophthalmol-2020-316450.
- Brodin-Sartorius A, TРte MJ, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int 2012;81(2):179-89.
- Hohenfellner K, Niel C, Haffner D, et al. Beneficial effects of starting oral cysteamine treatment in the first 2 months of life on glomerular and tubular kidney function in infantile nephropathic cystinosis. Mol Genet Metab 2022;136:282-8.
- Emma F, van’t Hoff W, Hohenfellner K, et al. An international cohort study spanning five decades assessed outcomes of nephropathic cystinosis. Kidney Int 2021; doi.org/10.1016/j.kint.2021.06.019.
- Manz F, Gretz N. Progression of chronic renal failure in a historical group of patients with nephropathic cystinosis: European Collaborative Study on Cystinosis. Pediatr Nephrol 1994;8:466–71.
- Cohen C, Charbit M, Chadefaux-Vekemans B, et al. Excellent long-term outcome of renal transplantation in cystinosis patients. Orphanet J Rare Dis 2015;10:90.
- Levtchenko E, Blom H, Wilmer M et al. ACE inhibitor enalapril diminishes albuminuria in patients with cystinosis. Clin Nephrol 2003;60:386–9.
- Emma F, Nesterova G, Langman C, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant 2014;29 Suppl 4(Suppl 4):iv87-iv94.