Болезнь Помпе у взрослых: клинические проявления, диагноз и лечение
Болезнь Помпе относится к редким наследственным лизосомным болезнями накопления и характеризуется накоплением гликогена в клетках мышц в результате снижения активности лизосомной кислой a-глюкозидазы. Симптомы болезни Помпе могут появиться в любом возрасте, в том числе пожилом. Они включают в себя медленно прогрессирующую проксимальную миопатию, сопровождающуюся умеренным увеличением активности креатинкиназы и других мышечных ферментов, и нарушение функции дыхания, связанное с поражением дыхательных мышц. Для подтверждения диагноза определяют активность a-глюкозидазы в сухих пятнах крови и проводят молекулярно-генетическое исследование. В статье обсуждается случай болезни Помпе с поздним началом, иллюстрирующий эффективность ферментозаместительной терапии рекомбинантным препаратом кислой a-глюкозидазы (алглюкозидазой альфа).
Моисеев С.В. Болезнь Помпе, или гликогеноз II типа, – это редкое наследственное заболевание, связанное со снижением активности лизосомного фермента кислой a-глюкозидазы в результате мутаций гена GAA и характеризующееся накоплением гликогена в лизосомах клеток, прежде всего скелетных мышц и миокарда [1]. В настоящее время известно более 300 мутаций гена GAA [2]. Заболевание передается по аутосомно-рецессивному типу и встречается с одинаковой частотой у мужчин и женщин. Классическая (инфантильная) форма болезни Помпе, описанная голландским патологоанатомом Johannes Pompe в 1932 г., развивается вскоре после рождения, проявляется поражением сердца (гипертрофическая кардиомиопатия), тяжелой миопатией и дыхательной недостаточностью и без лечения обычно приводит к смерти в течение первого года жизни. Выделяют также позднюю форму болезни Помпе, первые симптомы которой появ ляются в любом возрасте (иногда даже в пожилом или старческом) и нарастают значительно медленнее [2]. В отличие от инфантильной (младенческой) формы, при позднем начале болезни Помпе поражение сердца отсутствует, а основным симптомом является нарастающая мышечная слабость, которая начинается с проксимальных мышц нижних конечностей, а затем распространяется на мышцы туловища, верхних конечностей и дыхательные мышцы, что приводит к нарушению двигательной активности и развитию дыхательной недостаточности, требующей респираторной поддержки. Причиной сравнительно медленного прогрессирования болезни Помпе с поздним началом считают сохранение остаточной активности кислой a-глюкозидазы (<30%), которая при инфантильной форме заболевания практически отсутствует (<1%) [3]. В России первое наблюдение болезни Помпе с поздним началом опубликовали С.С. Никитин и соавт. в 2014 году [4]. Какова эпидемиология болезни Помпе?
Муружева З.М. Распространенность бо лезни Помпе в общей популяции считают равной 1:40000 [5,6]. На долю поздней формы приходится примерно 3/4 случаев заболевания, а на долю инфантильной – 1/4. Однако истинная распространенность болезни Помпе, особенно с поздним началом, пре вышает указанный показатель. Напри мер, по данным скрининга у новорожденных, в различных штатах США она варьировалась от 1:23000 до 1:10000 [7-10], а в Италии составила 1:18000 [11].
Моисеев С.В. С 2023 года в нашей стране у новорожденных проводится скрининг на 36 наследственных заболеваний, однако лизосомные болезни не входят в их число. Оправдан ли скрининг на болезнь Помпе?
Муружева З.М. Основными условиями скрининга у новорожденных являются наличие надежных методов диагностики соответствующего наследственного заболевания и, что самое главное, доступность эффективного лечения, которое следует начинать сразу после установления диагноза. В случае инфантильной формы болезни Помпе целесообразность cкрининга не вызывает большого сомнения, учитывая практически 100% смертность таких пациентов и возможность ферментозаместительной терапии (ФЗТ), которая при своевременном назначении позволяет улучшить исходы заболевания [11]. Выявление вариантов гена GAA, ассоциирующихся с поздним началом болезни Помпе, позволяет в будущем сократить срок до установления диагноза при появлении признаков миопатии. Однако необходимо учитывать и негативные аспекты скрининга, в том числе отсутствие четкой корреляции между генотипом и фенотипом, возможность отсутствия симптомов на протяжении десятилетий или всей жизни, трудность интерпретации результатов молекулярно-генетического исследования при наличии вариантов гена неопределенного значения.
Моисеев С.В. Заподозрить диагноз наследственного заболевания проще у детей или при появлении первых симптомов в детском или подростковом возрасте, особенно при наличии семейного анамнеза. У взрослых врачи в первую очередь исключают приобретенные заболевания, а семейный анамнез при аутосомнорецессивном типе наследования обычно мало информативен, хотя определенное значение может иметь этническая принадлежность пациента. Так, в некоторых странах/регионах распространены кровнородственные браки, повышающие вероятность гомозиготного носительства мутантного гена. Клиническую картину болезни Помпе у взрослого пациента иллюстрирует следующее наблюдение.
Муружева З.М. Пациент К., 45 лет обратился в Клинику ФГБНУ "Институт экспериментальной медицины" в декабре 2021 года с жалобами на слабость в мышцах, преимущественно нижних конечностей, трудности при ходьбе (неустойчивость, частые падения, не может подняться по лестнице) и перемене положения тела, невозможность удерживать голову в вертикальном положении, нарушение осанки, утомляемость, одышку при минимальной физической нагрузке и в положении лежа (спит в полусидячем положении), опущение век, дневную сонливость, эпизоды засыпания в положении сидя, невнятность речи, отеки ног.
В детском и подростковом возрасте активно занимался легкой атлетикой, затруднений при этом не испытывал. Первые симптомы заболевания появились в возрасте 37-38 лет, когда стал отмечать повышенную утомляемость, снижение работоспособности, общую слабость, которая не проходила даже после отдыха, боли в мышцах. С 41 года рецидивирующие внебольничные пневмонии, по поводу чего неоднократно проходил стационарное лечение (всего 9 госпитализаций за 4 года). В возрасте 42 лет в связи с тяжелым течением пневмонии проводилась механическая вентиляция легких с наложением трахеостомы на 1 мес. Примерно с того же возраста стала нарастать общая слабость, появились нечеткость речи и слабость в ногах. Пациенту стало трудно подниматься по лестнице. За год до обращения в клинику перенес COVID-19 тяжелого течения, после которого усилилась одышка, появлявшаяся при непродолжительной ходьбе и разговоре. Диагностирована хроническая обструктивная болезнь легких, дыхательная недостаточность 2 степени. Установлена 3 группа инвалидности. В связи с ночной гиповентиляцией проводилась неинвазивная вентиляция легких.
В связи с нарастающей мышечной слабостью в ногах и нарушением речи обследован в неврологическом отделении, где на основании электронейромиографии установлен диагноз хронической воспалительной демиелинизирующей полиневропатии. Проводи лась терапия преднизолоном в дозе 60 мг/сут с постепенным снижением до 15 мг/сут. Через 3 месяца пациент самостоятельно прекратил прием препарата в связи с отсутствием положительной динамики. В течение последнего года отмечает прогрессирующее ухудшение состояния. Из-за слабости в мышцах нижних конечностей не мог подниматься по лестнице и неоднократно падал. Стало трудно вставать с постели и менять положение тела. Наблюдались внезапные эпизоды засыпания в дневное время. Обсуждались диагнозы энцефалопатии и полирадикулопатии. При последнем обращении к неврологу заподозрена спинальная мышечная атрофия и рекомендовано проведение молекулярно-генетического исследования.
Семейный анамнез: мать умерла в возрасте 55 лет от инсульта. Причина смерти отца, страдавшего анкилозирующим спондилитом, в 40-летнем возрасте неизвестна.
При осмотре в клинике: сознание ясное, контактен, интеллектуально-мнестические функции не снижены. Менингеальных знаков нет. Диаметры правого и левого зрачков одинаковые. Фотореакция сохранена. Полуптоз обоих верхних век. Движения глазных яблок в полном объеме. Нистагма нет. Язык по средней линии. Макроглоссия. Оживление рефлексов орального автоматизма. Дизартрия и дисфония. Умеренная гипотрофия мышц плечевого и тазового пояса, задней группы мышц бедра. Псевдогипертофия мышц голеней. Тонус мышц снижен в проксимальных отделах. Парез сгибателей шеи до 3,5-4 баллов, разгибателей шеи до 2-3 баллов. Парез мышц проксимальных отделов верхних конечностей до 3,5-4 баллов, проксимальных отделов нижних конечностей – 3 балла. Дистально сила в конечностях – 5 баллов. Парез аксиальной мускулатуры: мышцы передней стенки живота – 2 балла, параспинальные мышцы – 2-3 балла. Гиперлордоз (чрезмерный прогиб поясничного отдела позвоночника кпереди). Сухожильные рефлексы с верхних и нижних конечностей симметрично снижены. Патологических знаков и чувствительных нарушений нет. Коор ди на торные пробы выполняет удовлетворительно с двух сторон. В позе Ромберга легкая неустойчивость. Походка переваливающаяся. Использует миопатические приемы при вставании из положения лежа и при укладывании на кровать. Вставание с корточек "лестничного" типа: чтобы подняться, опирается руками о пол, а затем поднимается, опираясь руками о колени (симптом Говерса). Антероколлис (симптом "свисающей головы"). Функции тазовых органов не нарушены. Проба с 6-минутной ходьбой прекращена через 2 мин из-за выраженной одышки.
Общий анализ крови без изменений. С-реактивный белок 13,8 мг/л (<5 мг/л). При биохимическом исследовании крови выявлено повышение активности креатинкиназы до 846 Ед/л (в норме <190 Ед/л), ЛДГ до 389 Ед/л (125-220 Ед/л), а также небольшое повышение активности АЛТ до 63 Ед/л (<42 Ед/л), АСТ до 86 Ед/л (<37 Ед/л) и &gama;-глутамилтранспептидазы до 75 Ед/л (<49 Ед/л). При ретроспективной оценке биохимических анализов крови пациента за последние 5 лет также отмечалось стойкое 1,5-2-кратное повышение активности печеночных аминотрансфераз. Активность креатинкиназы ранее не определяли.
При эхокардиографии стенка левого желудочка и межжелудочковая перегородка не утолщены, фракция выброса левого желудочка не снижена (62%). Определялись небольшая дилатация правых предсердия и желудочка и трикуспидальная регургитация 1 степени.
При спирометрии выявлено значительное снижение форсированной жизненной емкости легких (ФЖЕЛ) до 54% от должной без признаков бронхиальной обструкции (объем форсированного выдоха [ОФВ1] 87% от должного).
При игольчатой электромиографии (ЭМГ) определялись диффузные первично-мышечные изменения во всех исследованных мышцах верхних и нижних конечностей со вторичными реиннервационными изменениями.
Магнитно-резонансная томография (МРТ) головного мозга: единичные очаговые изменения вещества мозга, наиболее вероятно дистрофического характера. Признаки формирующегося "пустого" турецкого седла.
Компьютерная томография сосудов головного мозга: сужение правой позвоночной артерии (до 50%), атеросклеротические изменения интракраниальных сегментов позвоночных артерий. Картина замкнутого Виллизиева круга. Аневризм и сосудистых мальформаций не выявлено. Дегенеративно-дистрофические изменения шейного отдела позвоночника.
МРТ мышц тазового пояса и бедер: выраженная жировая дистрофия мышц (рис. 1).
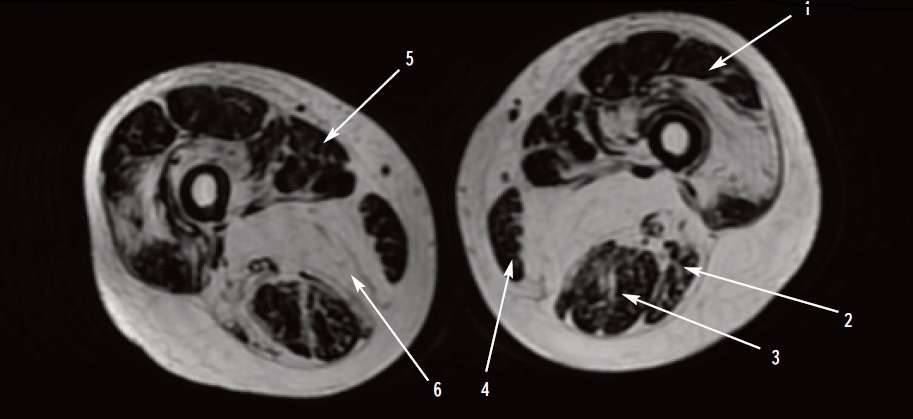
Таким образом, у 45-летнего пациента в течение примерно 7 лет наблюдалась прогрессирующая симметричная миопатия с поражением проксимальных мышц нижних и верхних конечностей и аксиальной мускулатуры. Рестриктивные нарушения вентиляции легких (нарастающая одышка, снижение ФЖЕЛ, повторные пневмонии) указывали на вовлечение в патологический процесс диафрагмы. Поражение последней может привести к снижению жизненной емкости легких в положении лежа, гиповентиляции в ночное время и сонливости в дневное время. При эхокардиографии признаков поражения миокарда не выявлено. Наличие миопатии подтверждалось данными игольчатой ЭМГ, МРТ мышц и повышением активности мышечных ферментов (креатинкиназа, ЛДГ, АСТ).
Моисеев С.В. Причинами проксимальной миопатии у взрослых могут быть наследственные, аутоиммунные и эндокринные заболевания, лекарственные средства, злокачественные опухоли, инфекционные агенты и др. Нарастающая мышечная слабость, скованность, судороги и замедление рефлексов, сопровождающиеся повышением активности креатинкиназы, часто наблюдаются при гипотиреозе и могут быть первыми проявлениями этого заболевания [12]. Ранние симптомы гипотиреоза неспецифичны, однако установить снижение функции щитовидной железы несложно при лабораторном исследовании (повышение активности тиреотропного гормона, снижение содержания гормонов щитовидной железы). Миопатия наблюдается и при гиперфункции щитовидной железы, а также гиперпаратиреозе и гиперкортицизме [12]. Важную роль в этиологии миопатии играют лекарственные средства, причем спектр проявлений лекарственного поражения мышц варьируется от легких миалгий или бессимптомного повышения активности креатинкиназы до тяжелой миопатии, характеризующейся прогрессирующей мышечной слабостью и атрофией мышц [13]. Чаще всего причиной лекарственной миопатии является мионекроз (например, при лечении статинами), который в тяжелых случаях может привести к рабдомиолизу, однако поражение мышц при медикаментозной терапии быть обусловлено и другими механизмами (табл. 1). В последние годы увеличивается значение ингибиторов конечных точек в этиологии иммуновоспалительных миопатий на фоне расширения показаний к их применению для лечения злокачественных опухолей. Если клинических и анамнестических данных в пользу вторичного генеза миопатии нет, как в нашем наблюдении, необходимо исключать не только наследственные заболевания, но и идиопатические воспалительные миопатии. Что следует учитывать при проведении дифференциального диагноза?
| Механизм развития | Препараты/вещества |
|---|---|
| Некротизирующая миопатия/рабдомиолиз | Статины, фибраты, алкоголь, героин |
| Иммуновоспалительные миопатии | Статины, a-интерферон, ингибиторы фактора некроза опухоли α, ингибиторы конечных точек, бевацизумаб |
| Митохондриальные миопатии | Антиретровирусные препараты, статины, клевудин |
| Лизосомные миопатии | Аминохинолиновые производные, амиодарон |
| Микротубулярные миопатии | Колхицин, винкристин |
| Миофибриллярные миопатии | Эметин |
| Катаболическая миопатия | Глюкокортикостероиды |
Новиков П.И. Идиопатические воспалительные миопатии – это неоднородная группа аутоиммунных заболеваний, характеризующихся хроническим воспалением мышц (миозит) и проявляющихся нарастающей мышечной слабостью и миалгиями. Поражение мышц нередко сочетается с различными внемышечными проявлениями, такими как лихорадка, изменения кожи, артрит, интерстициальное заболевание легких, синдром Рейно, дисфагия, миокардит. В настоящее время к идиопатическим воспалительным заболеваниям относят дерматомиозит, антисинтетазный синдром, иммуноопосредованную некротизирующую миопатию, миозит с включениями, полимиозит и миозит, сочетающийся с другими системными заболеваниями соединительной ткани, такими как системная красная волчанка, системная склеродермия, синдром Шегрена или ревматоидный артрит (перекрестный синдром) [14]. Своевременная диагностика идиопатических воспалительных миопатий имеет важное значение, учитывая эффективность терапии глюкокортикостероидами и иммуносупрессивными препаратами. Кроме того, дерматомиозит и другие воспалительные миопатии (за исключением антисинтетазного синдрома и миозита с включениями) ассоциируются с высоким риском злокачественных опухолей, которые могут быть выявлены как до, так и после установления диагноза миозита.
Для идиопатических воспалительных миопатий (за исключением миозита с включениями) в целом характерно острое или подострое начало заболевания, которое может привести к резкому ограничению двигательной активности в течение нескольких недель после появления первых симптомов. Предполагать воспалительный генез поражения мышц следует также при наличии клинических и лабораторных признаков воспаления, таких как лихорадка или повышение уровня С-реактивного белка (хотя они могут и отсутствовать), а также указанных выше системных проявлений. Типичные кожные проявления дерматомиозита включают в себя периорбитальную гелиотропную сыпь с отеком, эритему на лице, коленях, локтях, лодыжках, шее, на передней поверхности груди в виде буквы V, на спине и плечах (симптом "шали"), красные и розовые, иногда шелушащиеся узелки и бляшки на разгибательной поверхности пястно-фаланговых суставов (сыпь Готтрона) [15]. При компьютерной томографии у большинства пациентов с идиопатическими воспалительными миопатиями, прежде всего антисинтетазным синдромом, определяются интерстициальные изменения в легких, которые иногда быстро нарастают. Следует отметить, что, в отличие от наследственных миопатий, причиной дыхательной недостаточности при миозите является именно поражение интерстиция легких, а не дыхательных мышц. У 60% пациентов с идиопатическими воспалительными миопатиями определяются миозитспецифические антитела (анти-Jo1, анти-MDA5, анти-Mi2, анти-TIF-1g и др.), которые имеют не только диагностическое значение, но и ассоциируются с определенными клиническими фенотипами и риском злокачественных опухолей. Кроме того, могут быть выявлены аутоантитела, встречающиеся при других аутоиммунных заболеваниях, такие как антиRo52, анти-PM-Scl, анти-Ku и анти-U1RNP (миозит ассоциированные антитела). Важным методом диагностики является биопсия мышцы, которая позволяет подтвердить наличие воспаления, дифференцировать различные воспалительные миопатии и исключить другие варианты поражения мышц, хотя интерпретация гистологических данных может быть затруднительной.
Моисеев С.В. Отсутствие данных в пользу вторичной миопатии или идиопатических воспалительных миопатий в представленном наблюдении заставляли предполагать наличие наследственного заболевания с поражением мышечной ткани. На чем основана их диагностика?
Ларионова В.И. Поясноконечностный тип поражения мышц с вовлечением дыхательной мускулатуры, медленное прогрессирование миопатии, отсутствие поражения сердечной мышцы, умеренное повышение активности креатинкиназы, данные игольчатой ЭМГ и МРТ характерны для болезни Помпе с поздним началом. Чтобы подтвердить этот диагноз, определяют активность кислой a-глюкозидазы с помощью тандемной масс-спектрометрии и, в случае ее снижения, проводят молекулярно-генетическое исследование [16]. Для выполнения этих тестов сухие пятна крови пациента были отправлены в лабораторию наследственных болезней обмена веществ Медико-генетического научного центра им. академика Н.П. Бочкова. При анализе образцов выявлено значительное снижение активности кислой a-глюкозидазы до 0,54 мкМ/л/ч (в норме 1,00–25,00 мкМ/л/ч), а при молекулярно-генетическом исследовании — варианты нуклеотидной последовательности c.2662G>T в экзоне 19 и c.-32-13T>G в интроне 1 гена GAA в гетерозиготном состоянии. Полученные данные подтверждали диагноз болезни Помпе. Следует отметить, что большинство пациентов с этим заболеванием являются компаунд-гетерозиготными, т.е. имеют две различных патогенных мутации гена GAA.
При миопатии неясного генеза может быть выполнена биопсия мышцы. При болезни Помпе в биоптате определяется вакуолизация мышечной ткани за счет отложения гликогена, степень которой обычно коррелирует с выраженностью клинических проявлений. Однако изменения в биоптате могут отсутствовать или неспецифичны, а отложение гликогена в скелетных мышцах наблюдаются и при других заболеваниях. В связи с этим значение биопсии мышц в диагностике болезни Помпе ограничено [17], хотя ее результаты помогают исключить воспалительный характер миопатии.
Поражение мышц может быть обусловлено различными причинами. Например, в представленном наминаблюдении обсуждался диагноз спинальной мышечной атрофии (СМА) — нервно-мышечного заболевания, обусловленного мутациями гена SMN1,кодирующего белок выживаемости мотонейронов(SMN) [18]. СМА характеризуется прогрессирующейгибелью двигательных нейронов передних рогов спинного мозга и ядер черепных нервов ствола мозга и проявляется симметричными параличами и атрофиейскелетной мускулатуры, в первую очередь проксимальных отделов конечностей. Иногда первые симптомыСМА появляются в возрасте 20-30 лет, а заболеваниепрогрессирует медленно. При обследовании нередкоопределяется умеренное повышение активности креатинкиназы, которое служит основанием для ошибочнойдиагностики первичного поражения мышечной ткани.Важное диагностическое значение имеются результатыигольчатой ЭМГ, позволяющей выявить поражениемотонейронов и установить нейрогенный генез атрофии мышц.
Болезнь Помпе необходимо также дифференцировать с другими наследственными миопатиями, такими как мышечные дистрофии Дюшенна и Беккера, или дистрофинопатии, которые обусловлены мутациями гена DMD, кодирующего дистрофин — белок, который необходим для нормального функционирования мышечной ткани [19]. Тип наследования сцеплен с Х-хромосомой, поэтому заболевания проявляются в основном у мужчин, у которых мутантный ген находится в гемизиготном положении. При миодистрофии Дюшенна дистрофин практически не синтезируется, а симметричная слабость в тазовом поясе и проксимальных отделах нижних конечностей появляется в детском возрасте и быстро нарастает, в результате чего большинство пациентов утрачивают способность ходить к 12 годам. Нередко наблюдается гипертрофическая кардиомиопатия. При миодистрофии Беккера дистрофин продолжается синтезироваться, хотя и в недостаточном количестве или с измененной структурой, поэтому эта форма заболевания проявляется в старшем возрасте и характеризуется медленным прогрессированием и редким поражением сердца.
Поясноконечностная миодистрофия может быть также следствием мутаций нескольких десятков других генов (MYOT, LMNA, CAV3, DNAJB6, DES, TNPO3 и др.), которые передаются по доминантному или рецессивному типу [20]. Эти заболевания имеют сходный клинический фенотип, поэтому диагностика их часто возможна только при молекулярно-генетическом исследовании. Если клинические данные не позволяют ограничить список потенциальных причин миопатии, целесообразно проводить полноэкзомное секвенирование. В последние годы стоимость такого исследования снизилась и нередко оказывается сопоставимой со стоимостью анализа панели генов.
Моисеев С.В. Эффективным методом диагностики болезни Фабри, которая также относится к лизосомным болезням накопления, является скрининг в "группах риска", в которых вероятность ее выявления выше, чем в общей популяции (например, среди пациентов, получающих заместительную почечную терапию, перенесших инсульт в молодом возрасте или страдающих гипертрофией левого желудочка). Возможен ли скрининг на болезнь Помпе с поздним началом?
Ларионова В.И. В связи с неспецифичностью первых проявлений и медленным прогрессированием миопатии диагноз болезни Помпе часто устанавливают с большим опозданием, особенно у взрослых пациентов. Например, в нашем наблюдении заболевание было диагностировано только спустя 7-8 лет после начала миопатии, когда у пациента уже имелись инвалидизирующие проявления (резкое ограничение двигательной активности, тяжелая дыхательная недостаточность). При болезни Помпе, в отличие от многих других наследственных заболеваний мышц, доступно эффективное лечение рекомбинантным препаратом кислой a-глюкозидазы, позволяющее остановить или по крайней мере замедлить прогрессирование миопатии. В связи с этим при наличии у пациента миопатического синдрома неясного генеза оправдан скрининг на болезнь Помпе, тем более что все необходимые исследования в нашей стране проводятся бесплатно (для этого необходимо отправить сухие пятна крови курьерской службой в лабораторию МГНЦ им. академика Н.П. Бочкова). Для скрининга обычно определяют активность кислой α-глюкозидазы.
В многоцентровом исследовании, проводившемся в США, активность фермента измеряли у 906 пациентов в возрасте в среднем 52,5±15,8 лет со слабостью в проксимальных мышцах, повышением активности креатинкиназы и/или слабостью в мышцах шеи [21]. В случае снижения активности кислой a-глюкозидазы проводили молекулярно-генетическое исследование. Болезнь Помпе с поздним началом была диагностирована у 9 (1,0%) пациентов, у которых определялись низкая активность кислой a-глюкозидазы и две патогенных мутации гена GAA. Сходные данные были получены в итальянском исследовании, в котором среди более 1000 пациентов с аналогичными показаниями для скрининга болезнь Помпе была диагностирована в 1,6% случаев [22]. В похожем польском исследовании частота болезни Помпе с поздним началом составила 3,0% среди 337 пациентов в возрасте в среднем 32 года с проксимальной миопатией или стойким повышением активности креатинкиназы [23]. В крупном европейском исследовании, проводившемся в Великобритании и Германии, снижение активности кислой a-глюкозидазы в сухих пятнах крови было выявлено у 232 (7,6%) из 3076 пациентов с проксимальной миопатией и/или повышением активности креатинкиназы [24]. При молекулярно-генетическом исследовании диагноз болезни Помпе был подтвержден у 74 из них (2,4% от общего числа обследованных). У большинства пациентов заболевание проявлялось проксимальной миопатией, часто сочетавшейся с дыхательной недостаточностью, реже — изолированным повышением активности креатинкиназы.
Альтернативой измерению активности кислой a-глюкозидазы может быть молекулярно-генетическое исследование панели генов, ассоциирующихся с развитием миопатии. B. Nallamilli и соавт. анализировали мутации 35 таких генов, включая GAA, у 4656 детей и взрослых с поясноконечностной миопатией [25]. В целом мутации исследованных генов (прежде всего CAPN3, DYSF, FKRP и ANO5) были выявлены у 27% пациентов, а болезнь Помпе с поздним началом была диагностирована у 38 (0,8%). Как и в нашем случае, все пациенты были компаунд-гетерозиготами, а у 82% из них определялся вариант c.-32-13T>G гена GAA. Полученные данные представляют несомненный научный интерес, однако при их интерпретации необходимо учитывать, что у двух третей пациентов мутации исследованных генов выявлены не были, специфическое лечение диагностированных заболеваний (за исключением болезни Помпе) не существует, а затраты на молекулярно-диагностическое тестирование панели генов значительно превышают стоимость определения активности кислой &alha;-глюкозидазы.
Моисеев С.В. Гипертрофическая кардиомиопатия – типичное проявление инфантильной формы болезни Помпе. Возможно ли поражение сердца при поздней форме этого заболевания? Следует ли исключать болезнь Помпе у подростков и взрослых с гипертрофией левого желудочка неясного происхождения?
Ларионова В.И. В литературе описаны единичные случаи поражения сердца у пациентов с болезнью Помпе с поздним началом. H. Van Kooten и соавт. недавно проанализировали частоту нарушений сердечной деятельности у 750 таких больных на основании систематизированного обзора 48 исследований [26]. Частота гипертрофии левого желудочка или повышения индекса массы миокарда левого желудочка в разных исследованиях варьировалась от 4,6% до 16,7%. По мнению авторов, этот показатель соответствует таковому в общей популяции, а причиной утолщения стенки левого желудочка могли быть сопутствующие сердечнососудистые заболевания, прежде всего артериальная гипертония. В проанализированных исследованиях были зарегистрированы всего 3 случая гипертрофической кардиомиопатии, причем у всех пациентов наблюдалось очень раннее развитие заболевания. Кроме того, при болезни Помпе изменения со стороны сердца всегда сочетаются с мышечной слабостью и/или поражением дыхательной мускулатуры. Соответственно, исключать это заболевание у пациента с изолированной гипертрофией миокарда неясного генеза не имеет смысла.
Муружева З.М. В январе 2022 года у пациента развилась аспирационная пневмония, осложнившаяся тяжелой дыхательной недостаточностью, потребовавшей искусственной вентиляции легких. В апреле 2022 года была начата ФЗТ алглюкозидазой альфа в дозе 20 мг/кг один раз в 2 недели. После третьей инфузии пациент был переведен на самостоятельное дыхание, а после четвертой инфузии (1 июля) выписан домой. При выполнении теста с 6-минутной ходьбой пройденная дистанция составила 490 м (норма для пациента 453-606 м). Кроме того, пациент мог самостоятельно с односторонней поддержкой преодолеть лестничный пролет. Активность креатинкиназы снизилась до 254 Ед/л, а активность ЛДГ и печеночных аминотрансфераз нормализовалась. При осмотре в ноябре 2022 года, т.е. через 6 месяцев после начала ФЗТ, отмечено нарастание мышечной силы в разгибателях шеи до 3,5 баллов и в проксимальных мышцах верхних конечностей до 4,5-5 баллов. Пройденная за 6 минут дистанция увеличилась до 600 м. Пациент самостоятельно поднимается на второй этаж без поддержки.
Моисеев С.В. Представленное наблюдение иллюстрирует высокую эффективность ФЗТ у пациента с далеко зашедшей формой болезни Помпе. Каковы результаты клинических исследований алглюкозидазы альфа?
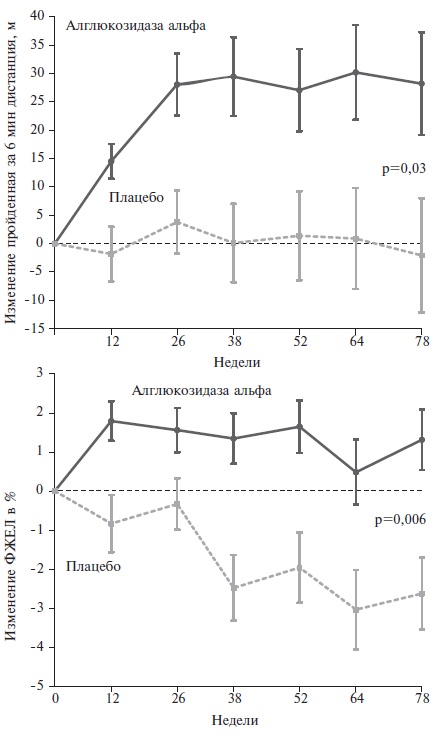
Муружева З.М. Препарат алглюкозидаза альфа был зарегистрирован в США и Европейском Союзе в 2006 году на основании результатов исследований у пациентов с инфантильной формой болезни Помпе. В 2010 году были опубликованы результаты многоцентрового двойного слепого, рандомизированного, плацебо-контролируемого исследования у 90 подростков (старше 8 лет) и взрослых с болезнью Помпе с поздним началом, которые в течение 78 недель получали алглюкозидазу альфа в дозе 20 мг/кг внутривенно каждые 2 недели или плацебо [27]. Первичными показателями эффективности были изменения пройденной за 6 минут дистанции и ФЖЕЛ.
По обоим показателям исследуемый препарат достоверно превосходил плацебо. Так, через 78 недель пройденная за 6 минут дистанция в основной группе увеличилась в среднем на 28,1±13,1 м (р=0,003), а ФЖЕЛ – на 3,4±1,2% (абсолютные изменения; р=0,006) по сравнению с плацебо (рис. 1). Улучшение отмечалось преимущественно в первые 26 недель, а эффективность ФЗТ была выше у пациентов с менее выраженными функциональными нарушения (исходные значения пройденной за 6 минут дистанции ≥300 м и ФЖЕЛ≥55% от должной). Показатели силы в мышцах верхних и нижних конечностей также увеличились, хотя их динамика не достигла статистической значимости.
Переносимость терапии была в целом хорошей. Основные побочные эффекты препарата включали в себя анафилактические реакции и инфузионные реакции (крапивница, приливы, потливость, дискомфорт в груди, рвота и повышение АД), которые наблюдались у 5-8% пациентов. У 2 из 3 пациентов, перенесших анафилактические реакции, лечение было прекращено. Таким образом, это исследование показало, что ФЗТ алглюкозидазой альфа вызывает увеличение переносимости физической нагрузки у пациентов с болезнью Помпе с поздним началом и стабилизирует функцию легких, которая при естественном течении болезни постепенно ухудшается (ежегодное снижение ФЖЕЛ на 1,7-4,6%) [28,29].
К настоящему времени опубликованы результаты длительных наблюдательных исследований, в которых эффективность ФЗТ у пациентов с болезнью Помпе с поздним началом оценивали в течение 5 и даже 10 лет [30-32]. Полученные данные подтвердили, что наиболее выраженный эффект алглюкозидазы альфа отмечается в течение первых нескольких месяцев после начала лечения, после чего наступает плато. Со временем у части пациентов несмотря на продолжение ФЗТ переносимость физической нагрузки и/или функция легких начинают постепенно ухудшаться, хотя в наиболее длительном исследовании даже через 10 лет после начала лечения более чем у половины пациентов пройденная за 6 минут дистанция и/или ФЖЕЛ оставались выше исходных значений [32].
Моисеев С.В. Недавно в разных странах, включая Российскую Федерации, был зарегистрирован новый препарат для лечения болезни Помпе – авалглюкозидаза альфа. Чем он отличается от алглюкозидазы альфа? Опубликованы ли результаты сравнительных исследований?
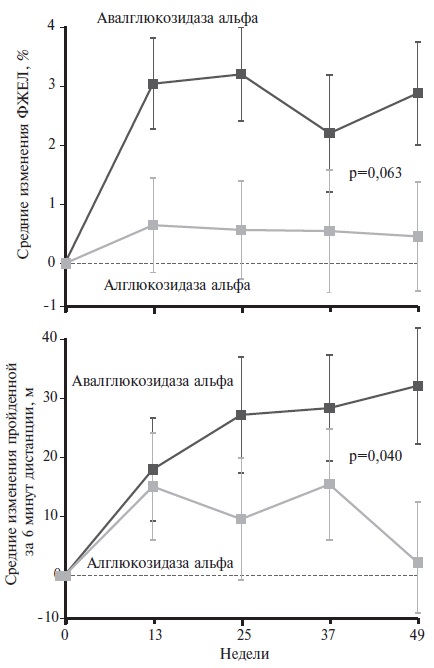
Муружева З.М. Интернализация алглюкозидазы альфа и ее транспорт в лизосомы опосредуются рецепторами маннозо-6-фосфата, которые находятся на поверхности клеток. По сравнению с алглюкозидазой альфа новая форма рекомбинантной кислой a-глюкозидазы содержит большее количество бис-маннозо-6фосфата (примерно в 15 раз), что обеспечивает лучшее проникнование препарата в клетки-мишени за счет увеличения его сродства к рецепторам. В доклинических исследованиях авалглюкозидаза альфа вызывала более значительное увеличение клиренса гликогена из клеток сердца, дыхательных и скелетных мышц и более выраженное улучшение двигательной функции по сравнению с алглюкозидазой альфа в эквивалентной дозе [33]. В 2021 году были опубликованы результаты двойного слепого, рандомизированного клинического исследования COMET, в котором эффективность алглюкозидазы альфа и авалглюкозидазы альфа сравнивали у 100 ранее нелеченных пациентов с поздней формой болезни Помпе (средний возраст 48,1±14,2 лет, диапазон от 16 до 78 лет) [34]. Целью исследования было доказать, что авалглюкозидаза альфа по влиянию на ФЖЕЛ по крайней мере не уступает алглюкозидазе альфа. Вторичным показателем эффективности служили изменения пройденной за 6 минут дистанции. Лечение авалглюкозидазой альфа через 49 недель привело к увеличению ФЖЕЛ в среднем на 2,89±0,88%, алглюкозидазой альфа – на 0,46±0,93%. При статическом анализе динамики этого показателя было установлено, что новый препарат по эффективности не уступает алглюкозидазе альфа, хотя преимущество авалглюкозидазы при статистическом анализе подтверждено не было (разница не достигла статистической значимости; р=0,063). Тем не менее, лечение авалглюкозидазой альфа привело к более значительному увеличению пройденной за 6 минут дистанции по сравнению с алглюкозидазой альфа – на 32,21±9,93 и 2,19±10,40 м, соответственно, а также силы в мышцах верхних и нижних конечностей (рис. 2). Все 5 случаев досрочного прекращения ФЗТ (в том числе 4 из-за нежелательных явлений) наблюдались в группе алглюкозидазы альфа.
Таким образом, результаты этого исследование показали, что длительная ФЗТ авалглюкозидазой альфа у пациентов с поздней формой болезни Помпе вызывает более выраженное улучшение функции легких, переносимости физической нагрузки и силы мышц, чем лечение алглюкозидазой альфа. Кроме того, новый препарат имел преимущества по профилю безопасности. В связи с этим мы планируем перевести нашего пациента на лечение авалглюкозидазой альфа, что может привести к улучшению результатов ФЗТ.
Моисеев С.В. Болезнь Помпе – это редкая наследственная лизосомная болезнь накопления, которая встречается не только у детей, но и взрослых. Основные проявления болезни Помпе с поздним началом – медленно нарастающая слабость в проксимальных мышцах нижних и верхних конечностей и туловища, сопровождающаяся умеренным повышением активности креатинкиназы и других мышечных ферментов, и дыхательная недостаточность. Выраженная гипертрофия левого желудочка наблюдается при инфантильной форме заболевания, развивающейся в первые месяцы после рождения, но практически не встречается при болезни Помпе с поздним началом. Учитывая неспецифичность проявлений болезни Помпе и доступность патогенетической терапии рекомбинантными препаратами кислой a-глюкозидазы, которая более эффективна на ранних стадиях заболеваний, целесообразно шире проводить скрининг на болезнь Помпе среди пациентов с проксимальной миопатией неясного происхождения путем определения активности кислой α-глюкозидазы и молекулярно-генетического исследования.
Используемые источники
- van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet. 2008;372(9646):1342–53.
- Stevens D, Milani-Nejad S, Mozaffar T. Pompe disease: a clinical, diagnostic, and therapeutic overview. Curr Treat Options Neurol 2022;24(11):573-88.
- Lim JA, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci 2014;6:177.
- Никитин С.С., Ковальчук М.О., Захарова Е.Ю., Цивилева В.В. Болезнь Помпе с поздним началом: первое клиническое описание в России. Нервно-мышечные болезни 2014;(1):62-8 [Nikitin SS, Kovalchuk MO, Zaharova EU, Tsivileva VV. Late-onset Pompe disease: first clinical description in Russia. Neuromuscular Diseases 2014;(1):62-8 (In Russ.)].
- Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999;7(6):713–6.
- Martiniuk F, Chen A, Mack A, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet 1998;79(1):69–72.
- Tang H, Feuchtbaum L, Sciortino S, et al. The first year experience of newborn screening for Pompe disease in California. Int J Neonatal Screen 2020;6(1):9.
- Burton BK, Charrow J, Hoganson GE, et al. Newborn screening for Pompe disease in Illinois: experience with 684,290 infants. Int J Neonatal Screen 2020;6(1):4.
- Ficicioglu C, Ahrens-Nicklas RC, Barch J, et al. Newborn screening for Pompe disease: Pennsylvania experience. Int J Neonatal Screen 2020;6(4):89.
- Klug TL, Swartz LB, Washburn J, et al. Lessons learned from Pompe disease newborn screening and follow-up. Int J Neonatal Screen 2020;6(1):11.
- Gragnaniello V, Pijnappel PWWM, et al. Newborn screening for Pompe disease in Italy: Long-term results and future challenges. Mol Genet Metab Rep 2022;33:100929.
- Rodolico C, Bonanno C, Pugliese A, et al. Endocrine myopathies: clinical and histopathological features of the major forms. Acta Myol 2020;39(3):130-5.
- Mastaglia FL. The changing spectrum of drug-induced myopathies. Acta Myol 2020;39:283-8.
- Lundberg IE, Fujimoto M, Vencovsky J, et al. Idiopathic inflammatory myopathies. Nat Rev Dis Primers 2021;7(1):86.
- Зыкова А.С., Новиков П.И., Моисеев С.В. Дерматомиозит взрослых: новые критерии диагностики и перспективы терапии. Клин фармакол тер 2017;26(2):83-92 [Zykova A, Novikov P, Moiseev S. Adult-onset dermatomyoisitis: new classification criteria and modern treatment. Clin Pharmacol Ther 2017;26(2):83-92 (In Russ.)].
- Erdem Ozdamar S, Koc AF, Durmus Tekce H, et al. Expert opinion on the diagnostic odyssey and management of late-onset Pompe disease: a neurologist's perspective. Front Neurol 2023;14:1095134.
- Савостьянов К.В., Никитин С.С., Карпачева К.Е. Лабораторные исследования и болезнь Помпе: от подозрения до мониторинга терапии. Нервномышечные болезни 2016;6(1):54-62 [Savost’yanov KV, Nikitin SS, Karpacheva KE. Laboratory studies and Pompe disease: from suspicion to therapy monitoring. Neuromuscular diseases 2016;6(1):54-62 (In Russ.)].
- Шпилюкова Ю.А., Иллариошкин С.Н. Спинальная мышечная атрофия у взрослых: проблемы ранней диагностики. Нервно-мышечные болезни 2022;12(4):37-45. Shpilyukova YuA, Illarioshkin SN. Adult spinal muscular atrophy: problems of early diagnosis. Nervno-myshechnye bolezni = Neuromuscular Diseases 2022;12(4):37-45. (In Russ.)].
- Китаева В.Е., Котов А.С., Бунак М.С. Прогрессирующие мышечные дистрофии. Российский неврологический журнал 2021;26(2):43-57 [Kitaeva VE, Kotov AS, Bunak MS. Progressive muscular dystrophies. Russian Neurological Journal = Rossijskij Nevrologicheskiy Zhurnal 2021;26(2):43-57 (In Russ.)].
- Bouchard C, Tremblay JP. Limb-girdle muscular dystrophies classification and therapies. J Clin Med 2023;12(14):4769.
- Wencel M, Shaibani A, Goyal NA, et al. Investigating late-onset Pompe prevalence in neuromuscular medicine academic practices: The IPaNeMA Study. Neurol Genet 2021;7(6):e623.
- Musumeci O, la Marca G, Spada M, et al. LOPED study: looking for an early diagnosis in a late-onset Pompe disease high-risk population. J Neurol Neurosurg Psychiatry 2016;87(1):5-11.
- Jastrzębska A, Potulska-Chromik A, Łusakowska A, et al. Screening for late-onset Pompe disease in Poland. Acta Neurol Scand 2019;140(4):239-43.
- Lukacs Z, Nieves Cobos P, et al. Prevalence of Pompe disease in 3,076 patients with hyperCKemia and limb-girdle muscular weakness. Neurology 2016;87(3):295-8.
- Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol 2018;5(12):1574-87.
- Van Kooten HA, Roelen CHA, Brusse E, et al. Cardiovascular disease in nonclassic Pompe disease: A systematic review. Neuromuscul Disord 2021;31(2):79-90.
- Van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010;362 (15):1396–406.
- Wokke JH, Escolar DM, Pestronk A, et al. Clinical features of late-onset Pompe disease: a prospective cohort study. Muscle Nerve 2008;38:1236-45.
- Van der Beek NA, Hagemans ML, Reuser AJ, et al. Rate of disease progression during long-term follow-up of patients with late-onset Pompe disease. Neuromuscul Disord 2009;19:113-7.
- Harlaar L, Hogrel JY, Perniconi B, et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology 2019;93:e1756–67.
- Kuperus E, Kruijshaar ME, Wens SCA, et al. Long-term benefit of enzyme replacement therapy in Pompe disease: a 5-year prospective study. Neurology 2017;89(23):2365–73.
- GüngЪr D, Kruijshaar ME, Plug I, et al. Impact of enzyme replacement therapy on survival in adults with Pompe disease: results from a prospective international observational study. Orphanet J Rare Dis 2013;8:49.
- Zhu Y, Jiang JL, Gumlaw NK, et al. Glycoengineered acid alpha-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther 2009;17:954–63.
- Diaz-Manera J, Kishnani PS, Kushlaf H, et al; COMET Investigator Group. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol 2021;20(12):1012-26.