Хроническая почечная недостаточность при болезни Фабри: общая выживаемость при использовании различных методов заместительной почечной терапии
Оценить выживаемость у пациентов с болезнью Фабри (БФ) в зависимости от вида заместительной почечной терапии, и определить роль диализного скрининга в ранней диагностике БФ у родственников пробандов.
В исследование включали взрослых (старше 18 лет) пациентов с подтвержденным диагнозом БФ. Терминальную стадию хронической почечной недостаточности (тХПН) диагностировали в соответствии с рекомендациями Научного общества нефрологов России (2016) и KDIGO (2012). В семьях пробандов с тХПН проводили обследование родственников, которые могли унаследовать мутантный ген.
У 50 (24,9%) из 201 пациентов с БФ диагностирована тХПН, в том числе у 48 (40.0%) из 120 мужчин и 2 (2,7%) из 81 женщин. Кумулятивная частота регистрации тХПН, рассчитанная методом Каплана-Майера, увеличивалась к возрасту 20-30 лет, а к возрасту 50 лет ожидаемая доля пациентов с тХПН составила 95%. Пяти из 50 больных с тХПН была выполнена трансплантация почки, в среднем, через 17 мес (диапазон от 7 до 70 мес) после начала лечения гемодиализом. Умерли 15 (33,3%) из 45 пациентов (все мужчины, медиана возраста на момент смерти 45 лет), получавших лечение диализом. Среди пациентов, которым проведена трансплантация почки, летальных исходов зарегистрировано не было. У 44 (88%) из 50 пациентов диагноз БФ установлен, в среднем, через 1 год (диапазон от 0 до 12 лет) после начала лечения программным гемодиализом при скрининге, проводившемся в российских диализных отделениях. У 89 (57%) из 156 обследованных родственников пробандов с тХПН диагностирована БФ, в том числе у 18 детей и подростков в возрасте до 18 лет. У 80,4% обследованных родственников пробандов с тХПН обнаружено поражение почек, преимущественно на ранних стадиях.
тХПН — нередкое осложнение БФ, ассоциированное с неблагоприятным прогнозом. Скрининг в диализных отделениях – эффективный способ выявления пробандов с БФ, открывающий возможность диагностики заболевания у их родственников на ранних стадиях, когда наиболее эффективна ферментозаместительная терапия.
Болезнь Фабри (БФ) – это орфанная наследственная лизосомная болезнь накопления. В основе патогенеза БФ лежит снижение или полное отсутствие активности лизосомного фермента α-галактозидазы А вследствие мутации гена GLA, расположенного на Х-хромосоме, что приводит к накоплению гликосфинголипидов, преимущественно деацитилированной фор мы глоботриаозилсфингозина (Lyso-GL3), в лизосомах клеток различных тканей и прогрессирующей дисфункции внутренних органов [1,2]. Сфинголипиды откладываются во всех структурах нефрона: в подоцитах, проксимальных и дистальных канальцах, перитубулярных капиллярах, в интиме и медии сосудов, что ведет к формированию гиалиноза артерий, прогрессирующему склерозу клубочков и интерстициальному фиброзу [3]. У большинства пациентов с БФ нефропатия проявляется альбуминурией, уровень которой с возрастом постепенно нарастает [4,5]. Наличие нефротического синдрома для пациентов с БФ не характерно. Ранним признаком повреждения почек при БФ может быть гиперфильтрация [6], а к возрасту 35-40 лет у пациентов с БФ отмечают прогрессирующее снижение скорости клубочковой фильтрации (СКФ) с формированием терминальной стадии хронической почечной недостаточности (тХПН) [7].
Важность своевременной диагностики БФ обусловлена возможностью ферментозаместительной терапии (ФЗТ) рекомбинантными препаратами α-галактозидазы А, которая позволяет задержать прогрессирующее повреждение органов [8]. По данным международного регистра Fabry Registry, средний срок от дебюта заболевания до установления диагноза БФ составляет 14 лет среди мужчин и 19 лет среди женщин [9]. С одной стороны, диагностика БФ затруднена наличием у части пациентов позднего фенотипа, характеризующегося отсутствием классических симптомов [9,10], с другой – многообразием клинических проявлений, что может создавать различные "маски" этого заболевания [11]. Пациенты с недиагностированной БФ могут быть выявлены путем скрининга в группах риска, например, среди больных с тХПН, получающих лечение диализом, гипертрофией левого желудочка неясного происхождения или инсультом, развившимся в молодом возрасте (до 60 лет). Подобные программы высоко затратны, так как частота диагностики БФ в указанных группах обычно не превышает 1%, т.е. чтобы диагностировать БФ у 1 больного, необходимо обследовать по крайней мере 100-200 пациентов или более. Тем не менее, скрининг в группах высокого риска открывает возможность семейного скрининга, позволяющего установить диагноз у родственников больных, в том числе детей, и своевременно начать ФЗТ.
Целью исследования было оценить выживаемость у пациентов с БФ в зависимости от вида заместительной почечной терапии (ЗПТ), и определить роль диализного скрининга в ранней диагностике БФ у родственников пробандов.
Материл и методы
В исследование включали взрослых (старше 18 лет) пациентов с БФ, обследованных в клинике им. Е.М. Тареева. Диагноз болезни Фабри устанавливали на основании наличия мутации гена GLA в сочетании с повышением концентрации Lyso-GL3 (у мужчин и женщин), снижением активности α-галактозидазы А в высушенных каплях крови (у мужчин), по крайней мере одним типичным проявлением БФ (нейропатическая боль, ангиокератомы, вихревидная кератопатия) и/или наличием определенного диагноза БФ у родственника [12]. У родственников пробанда диагноз болезни Фабри устанавливали на основании наличия такой же мутации гена GLA. Всех пациентов опрашивали, чтобы установить индексного пациента (пробанда) и его родственников, которые могли унаследовать мутантный ген с учетом Х-сцепленного типа наследования. При необходимости связывались с родственниками больного, чтобы получить более подробную информацию о семье. На основании собранной информации строили и анализировали родословные для каждого пробанда.
Молекулярно-генетическое исследование и определение содержания α-галактозидазы А и Lyso-GL3 проводили в лабораториях Centogene AG (Росток, Германия), ARCHIMED Life Science GmbH (Вена, Австрия), Медикогенетического научного центра имени академика Н.П. Бочкова и/или Национального медицинского исследовательского Центра Здоровья Детей. Активность α-галактозидазы А и уровень lyso-GL3 измеряли методом тандемной масс-спектрометрии в высушенных каплях крови. Нормальным считали содержание Lyso-GL3 менее 2,0 нг/мл. При молекулярно-генетическом исследовании изучали все кодирующие экзоны (1-7) с прилегающими интронными областями гена GLA. Минорные варианты генома с частотой менее 0,5% в соответствии с информационной базой gnomAD19, в том числе нуклеотидные варианты, не описанные ранее, подвергались биоинформатическому анализу в программе Alamut Visual (Interactive Biosoftware, Франция) и валидировались при помощи секвенирования по Сэнгеру. Последовательности нуклеотидов сравнивали с референсной базой данных GenBank Accession20 с использованием программы Geneious, версия R10 (Biomatters, Новая Зеландия).
У всех больных определяли суточную альбуминурию турбидиметрическим методом, протеинурию стандартным методом, сывороточный уровень креатинина и рассчи тывали СКФ по формуле CKD-EPI. Экскрецию альбумина/белка с мочой и функцию почек оценивали на основании рекомендаций KDIGO 2012 года и Научного общества нефрологов России 2016 года [13,14]. Почечным исходом считали наличие тХПН, требующей проведения ЗПТ в течение 6 и более месяцев. Очаговые изменения в белом вещества головного мозга и признаки перенесенного инсульта определяли при МРТ головного мозга (Siemens Magnetom Skyra 3 Тесла). МРТ сердца выполняли на магнитно-резонансных томографах с напряженностью поля 1,5 Тл Магнетом Аванто (Siemens Healthcare, Germany) или Оптима 450 (GE Healthcare, USA).
Статистический анализ проводили с использованием языка Python версии 3.9.12 (PSF, США) в среде разработки Jupyter Notebook. Нормальность распределения определяли с помощью критерия Шапиро-Уилка. Учитывая, что большинство признаков имели распределение, отличное от нормального, данные для количественных показателей, приведены в виде медианы и межквартильного размаха или диапазона. Данные для качественных переменных представлены в виде абсолютных частот и доли в процентах. Анализ выживаемости проводили с использованием метода Каплана-Майера (KaplanMeierFitter из библиотеки lifelines). Сравнение выживаемости по группам проводили с помощью лог-рангового теста. Различия считали значимыми при величине p<0,05.
Результаты
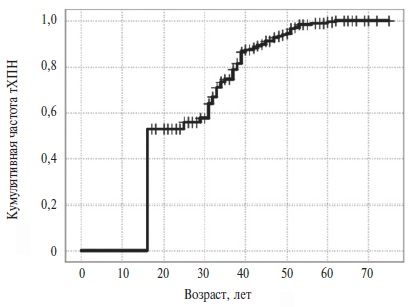
Клинико-демографическая характеристика пациентов. За период с 2010 по 2023 год в Клинике им. Е.М. Тареева обследовано 201 взрослых пациентов с БФ. Диализ зависимая тХПН диагностирована у 50 (24,9%) из 201 больных, в том числе у 48 (40%) из 120 мужчин и 2 (2,7%) из 81 женщин. У 42 (84%) из 50 пациентов почечный исход наступил раньше других тяжелых органных осложнений, таких как инсульт или клинически значимое нарушение ритма сердца. У остальных 8 (16%) пациентов первым зарегистрированным неблагоприятным исходом был инсульт. Медиана возраста начала ЗПТ составила 39 (33,8; 47,3) лет. Оценка кумулятивной частоты методом Каплана-Майера демонстрирует выраженное увеличение частоты регистрации тХПН к возрасту 20-30 лет, а к возрасту 50 лет ожидаемая доля пациентов с тХПН составляет 95% (рис. 1).
Пяти из 50 больных с тХПН была выполнена трансплантация почки, остальные 45 пациентов получали лечение программным гемодиализом. От инициации лечения гемодиализом до проведения трансплантации почки проходило, в среднем, 17 мес (диапазон от 7 до 70 мес). Срок наблюдения пациентов с БФ после трансплантации почки составил от 1 до 40 месяцев, уровень сывороточного креатинина после трансплантации – от 95 до 134 мкмоль/л.
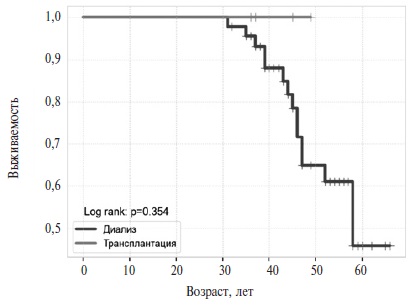
Умерли 15 (33,3%) из 45 пациентов, получавших лечение гемодиализом. Все умершие пациенты были мужского пола. Медиана возраста на момент летального исхода составила 45 (39; 58) лет. Среди пациентов, которым проведена трансплантация почки, летальных исходов зарегистрировано не было (рис. 2).
Результаты семейного скрининга. У 44 (88,0%) из 50 пациентов диагноз БФ установлен после начала лечения программным гемодиализом в процессе скрининга, проводившегося в российских диализных отделениях, в том числе у одного пациента – после трансплантации почки. Медиана срока от первых симптомов БФ до установки диагноза составила 24 (18; 36) года, а от начала лечения гемодиализом до установки диагноза БФ – 1 год (диапазон от 0 до 12 лет). У большинства пациентов (87%) с раннего возраста имелись классические проявления БФ, а на момент установки диагноза у большей части пациентов было выявлено поражение сердца и головного мозга (табл. 1).
| Параметры | Значения |
|---|---|
| Возраст, лет | 44,5 (35,0;52,0) |
| Мужчины, n (%) | 42 (95,5) |
| Lyso-Gl3, нг/мл | 70,1 (41,9;94,4) |
| Снижение активности α-галакатозидазы, n (%) | 42/44 (95,5) |
| Тип мутации, n (%) | |
| Миссенс | 24 (54,5) |
| Нонсенс | 8 (18,2) |
| Другие | 12 (27,3) |
| Классический фенотип, n (%) | 41 (87,2) |
| Ранние симптомы, n (%) | |
| Нейропатическая боль | 30 (68,2) |
| Ангиокератомы | 15 (34,1) |
| Гипо-/ангидроз | 31 (70,5) |
| Желудочно-кишечные проявления, n (%) | 9 (20,5) |
| Поражение органа зрения, n (%) | |
| Вихревидная кератопатия | 16/29 (55,2) |
| Катаракта Фабри | 6/29 (20,7) |
| Поражение сердца, n (%) | |
| Гипертрофия миокарда | 40/43 (93,0) |
| Нарушения ритма | 5 (11,4) |
| Поражение головного мозга, n (%) | |
| Очаги на МРТ головного мозга | 29/38 (76,3) |
| Инсульт | 13 (29,5) |
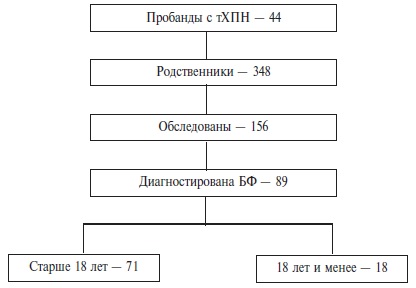
При опросе 44 пробандов с тХПН было установлено наличие 348 родственников (в 1-4 поколениях), которые могли быть носителями мутантного гена (рис. 3). Количество родственников на каждого пробанда составило, в среднем, 7 человек и варьировалось от 1 до 26 человек. У 156 (44,8%) родственников проведено обследование на БФ, включившее определение активности α-галактозидазы А у мужчин, содержаниия Lyso-GL3 и молекулярно-генетическое исследование у мужчин и женщин. В остальных случаях исследования не проводили в связи с отсутствием контактов внутри семьи (n=138) или отказом от тестирования (n=21). БФ диагностирована у 89 (57%) из 156 обследованных родственников, из которых 18 пациентов были детьми в возрасте до 18 лет. Среднее количество родственников на 1 пробанда составило 1,6 человек.
У 37 (80,4%) из 46 обследованных родственников диализных пробандов отмечено поражение почек. Медиана сывороточного креатинина у родственников составила 84,0 (65,5; 104,6) мкмоль/л, медиана суточной альбуминурии – 72,0 (33,6; 256,0) мг. У 82,6% родственников с нефропатией расчетная СКФ превышала 60 мл/мин/1,73 м2.
Обсуждение
тХПН является неизбежным исходом прогрессирующего поражения почек при естественном течении БФ. В нашем исследовании тХПН выявлена у 50 (24,9%) из 201 пациентов с БФ, в том числе у 40,0% мужчин и 2,7% женщин. Высокая распространенность тХПН в нашей выборке отчасти обусловлена тем, что у 88% из 50 больных диагноз БФ был установлен в результате скрининга, проводившегося в российских гемодиализных отделениях [15]. Несмотря на преобладание в нашей выборке пациентов (87%) с ранними классическими симптомами БФ, диагноз был установлен, в среднем, только через 24 года от дебюта болезни и через 1 год после начала заместительной почечной терапии. Поздняя диагностика демонстрирует существующие трудности первичного выявления пациентов с БФ. В связи с этим, для выявления пробандов остается необходимым скрининг в группах риска, в частности среди пациентов с тХПН неустановленной этиологии. Более того, при инициации лечения гемодиализом скрининг на БФ необходимо проводить в ранние сроки, учитывая высокую смертность пациентов. Ранее мы показали, что срок наступления летального исхода у пациентов с БФ после начала лечения гемодиализом составляет 32 мес [16]. К настоящему моменту в нашей выборке летальный исход зарегистрирован у 33,3% пациентов с БФ, получавших лечение гемодиализом, в то время как среди пациентов, которым проведена трансплантация почки, летальных исходов не было. Отсутствие статистических различий в смертности между двумя группами обусловлено небольшим количеством пациентов, перенесших трансплантацию почки.
В настоящее время трансплантация почки является оптимальным методом ЗПТ, обеспечивая наилучшую выживаемость пациентов, при этом прогноз улучшается при выполнении ранней трансплантации [17]. В отечественном исследовании, включившем 1197 пациентов c различными заболеваниями, находившихся в листе ожидания трансплантации почки, продемонстрировано увеличение смертности реципиентов почки при сроке ожидания трансплантации более 6 лет [18]. Срок ожидания трансплантации почки в нашей выборке пациентов с БФ не превышал 6 лет. К настоящему времени накоплен достаточно большой опыт трансплантации почки у пациентов с БФ. По данным Fabry Registry трансплантация почки была выполнена у 116 (62%) пациентов с БФ, нуждающихся в проведении ЗПТ [7]. Отдаленные результаты трансплантации почки у пациентов с БФ удовлетворительные. В исследовании T. Shah и соавт. пятилетняя выживаемость почечного трансплантата была выше среди 197 пациентов с БФ, чем среди более 200 тыс. пациентов с другими причинами тХПН (74% и 69%, соответственно, р=0,03) [19]. По данным метаанализа, включившего 424 реципиента почки с БФ, частота возврата заболевания в трансплантате составила 11,1%, а риск смерти от всех причин был сопоставим среди пациентов с БФ и пациентов с другими причинами тХПН [20]. По мнению экспертов, наличие тХПН или почечного трансплантата не является основанием для отказа от начала или продолжения ФЗТ, учитывая ее благоприятное влияние на другие проявления заболевания, такие как нейропатическую боль или гипертрофию миокарда левого желудочка, а также общий прогноз заболевания [8,21–23]. Кроме того, иммуносупрессивная терапия, назначаемая после трансплантации, может оказывать протективное влияние в отношении формирования антител к рекомбинантной агалсидазе А, которое может сопровождаться снижением эффективности ФЗТ [24]. В исследовании, включившем 24 реципиента почки с БФ со средним сроком наблюдения после трансплантации 80 мес и средним сроком лечения ФЗТ 9,8 лет, продемонстрирован благоприятный эффект иммуносупрессивной терапии на формирование антител к ФЗТ, что потенциально может повышать ее эффективность [25].
дения скрининга в группах высокого риска обоснована возможностью не только выявления пациентов с БФ с тяжелым поражением внутренних органов, но и диагностики заболевания у родственников пробандов, в том числе детей, на ранних стадиях заболевания. По данным нашего исследования, более половины из обследованных родственников пробандов с тХПН были носителями мутантного гена, а клинические проявления БФ имелись у трети из них. На каждого пробанда приходилось в среднем почти два новых пациентов с БФ. Следует отметить, что около половины родственников в нашей выборке не были обследованы. Основной причиной были плохие внутрисемейные связи и отсутствие общения, что часто было связано с географическим фактором. По данным семейного скрининга, проведенного в других странах, количество родственников с БФ, выявляемых на каждого пробанда, было выше и достигало 10 человек [26,27]. При этом у большинства родственников отмечались малосимптомное течение БФ и отсутствие тяжелых органных поражений. По нашим данным у 80,4% родственников пробандов с тХПН подтверждено вовлечение почек, однако у большей части – на ранних стадиях заболевания.
Кроме групп пациентов с тяжелыми органными поражениями, такими как тХПН, инсульт и гипертрофическая кардиомиопатия, развившимися в молодом возрасте, другой возможной группой риска могут быть пациенты с нефропатией неясной этиологии [28]. Так, в Турции были обследованы 53 пациента с нефропатией неустановленной этиологии без нефротического синдрома, у которых при биопсии почки со световой микроскопией нефробиоптатов, наблюдались вакуолизация клеток, фокальный и/или сегментарный гломерулосклероз и тубулоинтерстициальный фиброз [29]. Снижение активности α-галактозидазы А и патогенную мутацию в гене GLA выявили у 2 (5,4%) из 37 мужчин, в то время как у женщин патогенные варианты гена отсутствовали. У обоих мужчин с БФ при биопсии почки был диагностирован фокальный сегментарный гломерулосклероз. Среди всех пациентов, перенесших биопсию почки, распространенность БФ оказалась ниже – 5 (0,83%) из 600 пациентов [30], хотя она была сопоставимой с таковой в других группах риска, например, в отделениях гемодиализа – 20 (0,36%) из 5572 пациентов [15]. Достоверно установить морфологический диагноз БФ можно только с помощью электронной микроскопии нефробиоптатов, позволяющей выявить "зебровидные" включения и "миелиновые" тельца в цитоплазме всех клеток почки. Однако в российских нефрологических отделениях электронная микроскопия биоптатов почки проводится далеко не всегда. Исходя из вышесказанного, представляется целесообразным проведение скрининга на БФ среди пациентов с морфологически верифицированным фокальным сегментарным гломерулосклерозом неустановленной этиологии без нефротического синдрома.
Важность своевременной диагностики БФ обусловлена возможностью проведения ФЗТ, которая позволяет задержать прогрессирующее повреждение органов. В нашей выборке с учетом задержки в диагностике средний возраст пациентов на момент инициации ФЗТ составил 38 лет, а у части пациентов к моменту начала лечения уже имелись необратимые изменения внутренних органов [31]. При естественном течении БФ темп снижения СКФ у пациентов с высокой протеинурией достигает 5 мл/мин/1,73 м2 в год [7,32]. ФЗТ замедляет темп снижения СКФ, особенно если лечение начинают в молодом возрасте [8]. По нашим ранее опубликованным данным, среди пациентов с БФ, получавших ФЗТ (медиана длительности наблюдения – 5 лет), темп снижения СКФ составил, в среднем, 3 мл/мин/1,73 м2 в год. У мужчин он был выше, чем у женщин: 4,5 и 2,4 мл/мин/1,73 м2 в год, соответственно [31]. D. Germain и соавт. проанализировали частоту неблагоприятных исходов БФ, в том числе тХПН, у 52 пациентов, получавших ФЗТ агалсидазой бета в течение 10 лет. В этом исследовании выживаемость составила 94%, а у 81% больных в течение указанного срока не были зарегистрированы какие-либо тяжелые клинические исходы [33]. Эффективность ФЗТ в значительной степени зависит от срока ее назначения после появления первых симптомов и исходного состояния почек. Более поздняя терапия, особенно у пациентов с протеинурией и/или снижением СКФ, в меньшей степени влияет на течение нефропатии Фабри [34].
Ограничением нашего исследования является небольшая выборка пациентов, перенесших трансплантацию почки и короткий срок наблюдения за ними, что не позволило достоверно оценить выживаемость этих пациентов. Тем не менее, мы изучили выживаемость пациентов у достаточно большой группы диализных больных, а также провели семейный скрининг у всех пробандов с тХПН и оценили выраженность вовлечения почек у их родственников с подтвержденной БФ.
Заключение
тХПН – нередкое осложнение БФ, ассоциированное с неблагоприятным прогнозом. Улучшение прогноза у этих пациентов возможно при ранней диагностике БФ и проведении трансплантации почки с одновременным назначением патогенетической ФЗТ. Скрининг в группах высокого риска, таких как пациенты диализных отделений или с морфологически верифицированным фокальным сегментарным гломерулосклерозом неустановленной этиологии без нефротического синдрома, по-прежнему остается эффективным способом выявления пробандов с БФ. Более того, скрининг в группах риска открывает возможность семейного скрининга, позволяющего установить диагноз у родственников пробандов на ранних стадиях, когда лечение наиболее эффективно. Обследование родственников повышает эффективность затрат на скрининг в группах риска.
Используемые источники
- Zarate YA, Hopkin RJ. Fabry’s disease. Lancet 2008;372(9647):1427–35.
- Моисеев С.В., Тао Е.А., Моисеев А.С. и др. Клинические проявления и исходы болезни Фабри у 150 взрослых пациентов. Клин фармакол тер 2021;30(3):43-51 [Moiseev S, Tao E, Moiseev A, et al. Clinical manifestations and outcomes of Fabry disease in 150 adult patients. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(3):43-51 (In Russ.)].
- Fogo AB, Bostad L, Svarstad E, и др. Scoring system for renal pathology in Fabry disease: Report of the International Study Group of Fabry Nephropathy (ISGFN). Nephrol Dial Transplant 2010;25(7):2168–77.
- Ortiz A, Oliveira JP, Waldek S, et al. Nephropathy in males and females with Fabry disease: Cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant 2008;23(5):1600–7.
- Каровайкина Е.А., Моисеев С.В., Буланов Н.М. и др. Распространенность и основные проявления поражения почек у пациентов с болезнью Фабри. Клин фармакол тер 2018;27(4):46–52. Karovaikina E, Moiseev S, Bulanov N, et al. Prevalence and clinical manifestations of nephropathy in patients with Fabry disease. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2018;27(4):46–52 (In Russ.)].
- Riccio E, Sabbatini M, Bruzzese D, et al. Glomerular hyperfiltration: an early marker of nephropathy in Fabry disease. Nephron 2019;141(1):10–7.
- Ortiz A, Cianciaruso B, Cizmarik M, et al. End-stage renal disease in patients with Fabry disease: Natural history data from the Fabry Registry. Nephrol Dial Transplant 2009;25(3):769–75.
- Kampmann C, Perrin A, Beck M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: cardiac outcomes after 10 years’ treatment. Orphanet J Rare Dis 2015;10(1):21–7.
- Eng CM, Fletcher J, Wilcox WR, et al. Fabry disease: Baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis 2007;30(2):184–92.
- Nakao S, Kodama C, Takenaka T, et al. Fabry disease: Detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int 2003;64(3):801–7.
- Moiseev S, Karovaikina E, Novikov PI, et al. What rheumatologist should know about Fabry disease. Ann Rheum Dis 2020;79(6):e71.
- Smid BE, Van Der Tol L, Cecchi F, et al. Uncertain diagnosis of Fabry disease: Consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int J Cardiol 2014;177:400–8.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int Suppl 2012;3(1):1–150.
- Шилов Е.М., Смирнов А.В., Козловская Н.Л. Нефрология. Клинические рекомендации; 2016.
- Moiseev S, Fomin V, Savostyanov K, et al. The prevalence and clinical features of Fabry disease in hemodialysis patients: Russian Nationwide Fabry Dialysis Screening Program. Nephron 2019;141(4):249–55.
- Каровайкина Е.А., Моисеев С.В., Буланов Н.М. и др. Клинические проявления и исходы болезни Фабри у пациентов с терминальной стадией хронической почечной недостаточности. Нефрология и диализ 2019;21(1):72–7 [Karovaikina E, Moiseev S, Bulanov N, et al. Clinical manifestations and outcomes of Fabry disease in patients with terminal renal failure. Nephrology and dialysis 2019;21(1):72–7 (In Russ.)].
- Reese PP, Shults J, Bloom RD, et al. Functional status, time to transplantation, and survival benefit of kidney transplantation among wait-listed candidates. Am J Kidney Dis 2015;66(5):837–45.
- Ватазин А.В., Зулькарнаев А.Б., Степанов В.А. Анализ выживаемости пациентов в листе ожидания трансплантации почки с позиции конкурирующих рисков. Вестник трансплантологии и искусственных органов 2019;21(1):35–45 [Vatazin AV, Zulkarnaev AB, Stepanov VA. Survival analysis of patients in the waiting list for kidney transplantation in terms of competing risks. Russian Journal of Transplantology and Artificial Organs 2019;21(1):35-45 (In Russ.)].
- Shah T, Gill J, Malhotra N, Takemoto SK, Bunnapradist S. Kidney transplant outcomes in patients with Fabry disease. Transplantation 2009;87(2):280–5.
- Suarez MLG, Thongprayoon C, Hansrivijit P, и др. Outcomes of kidney transplantation in Fabry disease: A meta-analysis. Diseases 2020;9(1):2.
- Talbot AS, Lewis NT, Nicholls KM. Cardiovascular outcomes in Fabry disease are linked to severity of chronic kidney disease. Heart 2015;101(4):287–93
- Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab 2018;124(3):189–203.
- Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease. Ann Intern Med 2007;146(2):77–86.
- Deegan PB. Fabry disease, enzyme replacement therapy and the significance of antibody responses. J Inherit Metab Dis 2012;35(2):227–43.
- Lenders M, Oder D, Nowak A, et al. Impact of immunosuppressive therapy on therapy-neutralizing antibodies in transplanted patients with Fabry disease. J Intern Med 2017;282(3):241–253.
- Laney DA, Fernhoff PM. Diagnosis of Fabry disease via analysis of family history. J Genet Couns 2008;17(1):79–83.
- Rozenfeld PA, Masllorens FM, Roa N, et al. Fabry pedigree analysis: A successful program for targeted genetic approach. Mol Genet Genomic Med 2019;7(7):1–4.
- Schiffmann R, Hughes DA, Linthorst GE, et al. Screening, diagnosis, and management of patients with Fabry disease: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int 2017;91(2):284–93.
- OruН A, Yildiz A, AkgЯr S, et al. Screening for Fabry disease in patients who underwent renal biopsy and identification of a novel mutation. Turkish J Nephrol 2021;30(2):165–70.
- Лернер Ю.В., Цой Л.В., Гришина А.Н., Варшавский В.А. Морфологическая характеристика изменений почек при болезни Фабри. Архив патологии 2022;84(1):21–6 [Lerner YuV, Tsoy LV, Grishina AN, Varshavsky VA. Morphological characteristics of renal changes in Fabry disease. Archive of Pathology = Arkhiv patologii. 2022;84(1):21–26 (In Russ.)].
- Моисеев А.С., Буланов Н.М., Тао Е.А. и др. Эффективность и безопасность длительной ферментозаместительной терапии агалсидазой альфа и агалсидазой бета у взрослых пациентов с болезнью Фабри. Клин фармакол тер 2022;31(4):28-34 [Moiseev A, Bulanov N, Tao E, et al. Efficacy and safety of long-term enzyme replacement therapy with agalsidase alfa or agalsidase beta in adult patients with Fabry disease. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2022;31(4):28-34 (In Russ.)].
- Wanner C, Oliveira JP, Ortiz A, и др. Prognostic indicators of renal disease progression in adults with fabry disease: Natural history data from the Fabry Registry. Clin J Am Soc Nephrol 2010;5(12):2220–8.
- Germain DP, Charrow J, Desnick RJ, и др. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet 2015;52(5):353–8.
- Warnock DG, Ortiz A, Mauer M, и др. Renal outcomes of agalsidase beta treatment for Fabry disease: Role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant 2012;27(3):1042–9.