Атипичный гемолитико-уремический синдром: клиническая картина, диагностика и лечение
Атипичный гемолитико-уремический синдром (аГУС), или комплемент-опосредованная тромботическая микроангиопатия, – это редкое заболевание, обусловленное неконтролируемой активацией системы комлемента и характеризующееся тромбообразованием в сосудах микроциркуляторного русла и развитием острого повреждения почек в сочетании с тромбоцитопенией и микроангиопатической гемолитической анемией. Примерно у трети пациентов с аГУС наблюдается поражение не только почек, но и других органов, в частности центральной нервной системы, легких, печени, желудочно-кишечного тракта. Триггером развития или прогрессирования аГУС часто служат различные комплемент-активирующие состояния, в частности осложнения беременности (акушерский аГУС). Патогенетическая терапия аГУС предполагает проведение плазмотерапии и применение экулизумаба, блокирующего С5 компонент комплемента. В статье на основании двух наблюдений обсуждаются клинические проявления, диагностика, дифференциальная диагностика аГУС и подходы к его лечению.
С.В. Моисеев. Атипичный гемолитикоуре мический синдром (аГУС) – это редкое (орфанное) заболевание, несколько чаще развивающееся в детском или молодом возрасте [1,2]. Распространенность аГУС в популяции составляет от 2 до 9 случаев на 1 млн населения, а заболеваемость – от 0,23 до 1,9 на 1 млн [3]. аГУС представляет собой один из вариантов тромботической мик роангиопатии (ТМА) – клинико-морфологического синдрома, характеризующегося повреждением эндотелия, накоплением аморфного материала в субэндотелиальном пространстве и образованием тромбов в сосудах микроциркуляторного русла, что приводит к их окклюзии и ишемии органов и тканей, прежде всего почек [4]. Если не проводится патогенетическая терапия, то прогноз при аГУС неблагоприятный. В международном когортном исследовании через 3 года после первого эпизода ТМА умерли или нуждались в заместительной почечной терапии 79% из 273 пациентов с аГУС [5].
В основе патогенеза аГУС лежит неконтролируемая активация системы комплемента, изучение роли которой позволило значительно улучшить результаты лечения этого заболевания путем применения экулизумаба – рекомбинантного гуманизированного моноклонального антитела к С5 компоненту комплемента. Эффективность терапии выше, если ее назначают на более раннем этапе развития аГУС, что определяет важность своевременной диагностики заболевания. Как сегодня классифицируют аГУС? Какие еще заболевания могут быть причиной ТМА?
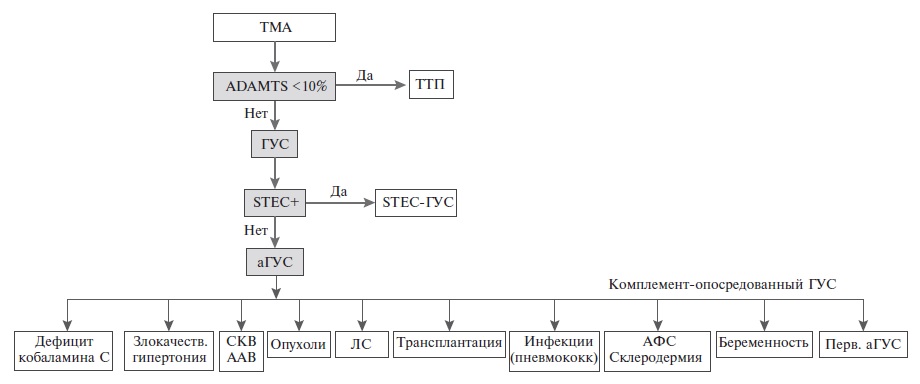
Н.Л. Козловская. Современная классификация ТМА выделяет первичные и вторичные ее формы. Первичная ТМА включает в себя тромботическую тромбоцитопеническую пурпуру (ТТП), типичный ГУС и аГУС. ТТП развивается в результате генетически обусловленного или, чаще, приобретенного (антитела, прием тиклопидина или клопидогрела) снижения активности ADAMTS-13 (менее 10%) – специфической протеазы, расщепляющей фактор Вилле бран да, увеличение содержания которого приводит к образованию тромбов в мелких сосудах. Типичный ГУС, сегодня получивший название STEC-ГУС, чаще всего наблюдается у детей и обусловлен прямым действием шига-токсина Escherichia coli, в то время как аГУС, или комплементопосредованная ТМА, развивается вследствие мутаций генов регуляторных белков или компонентов комплемента или образования антител к фактору Н системы комплемента. Эти изменения приводят к неконтролируемой активации системы комплемента, которая вызывает повреждение эндотелиальных клеток сосудов микроциркуляторного русла и последующее тромбообразование. Микрососуды почек наиболее уязвимы к активации системы комплемента, что обусловлено особым строением (фенестрация) эндотелия гломерулярных капилляров.
Традиционно выделяют семейный и спорадический аГУС. На долю первого, который диагностируют по крайней мере у двух членов семьи, приходится всего 1020% случаев аГУС. При спорадическом аГУС семейный анамнез отсутствует, хотя это не исключает наследственный характер заболевания, учитывая возможность носительства патогенных мутаций генов комплемента у родственников больного [6]. Сегодня некоторые исследователи предлагают также выделять первичный аГУС, обусловленный генетическими дефектами альтернативного пути активации комплемента, и вторичный ГУС, также ассоциированный с избыточной активацией комплемента, но опосредованный другими заболеваниями и состояниями, в том числе инфекциями, аутоиммунными заболеваниями, беременностью, опухолями, медикаментозной терапией, трансплантацией органов и др. (рис. 1) [7, 8]. Указанные факторы вызывают дополнительную активацию системы комплемента (комплемент-активирующие состояния) и выступают в роли триггеров, т.е. могут способствовать развитию или прогрессированию аГУС у предрасположенных пациентов, у которых имеются генетические дефекты системы комплемента. В связи с этим разделение аГУС на первичный и вторичный в значительной степени условно [9].
С.В. Моисеев. Каковы клинические проявления аГУС? Когда следует подозревать этот диагноз?
К.А. Демьянова. Для аГУС, как и любой другой ТМА, характерна триада клинических проявлений – тромбоцитопения, микроангиопатическая гемолитическая анемия (МАГА) и острое повреждение почек (ОПП). Тромбоцитопения (<150 × 109/л или снижение количества тромбоцитов по крайней мере на 25% от исходного) развивается в результате потребления тромбоцитов в процессе тромбообразования в сосудах микроциркуляторного русла. Причиной МАГА является механический гемолиз эритроцитов, обусловленный их повреждением при контакте с тромбами, отражением которого является увеличение количества фрагментированных эритроцитов, называемых шистоцитами (>0,1%). Другие проявления внутрисосудистого гемолиза включают в себя Кумбс-негативную анемию, повышение активности ЛДГ и снижение содержания гаптоглобина – a2-глобулина плазмы крови, связывающего гемоглобин [10]. Усиленное разрушение эритроцитов в кровяном русле приводит к увеличению поступления гемоглобина в кровь и, соответственно, к снижению содержания гаптоглобина, так как его образование при гемолизе не увеличивается. У всех больных с аГУС наблюдается поражение почек, в большинстве случаев проявляющееся олигурическим ОПП и примерно у 2/3 больных приводящее к развитию терминальной хронической почечной недостаточности. Большинство пациентов с аГУС при госпитализации в стационар уже нуждаются в заместительной почечной терапии. Характерна также артериальная гипертония, которая обусловлена увеличением объема циркулирующей крови на фоне олигурии/анурии и гиперренинемией, связанной с ишемией почечной ткани. При сохранении диуреза обычно определяется протеинурия, в том числе нефротического уровня, а также гематурия. Примером может служить следующее наблюдение.
А.В. Скворцов. Пациент Т., 26 лет. В начале января 2019 г. на фоне переохлаждения и правосторонней пневмонии отмечено прогрессирующее ухудшение состояния с развитием ОПП (анурия, увеличение сывороточного уровня креатинина до 1100 мкмоль/л, гипергидратация), которое сопровождалось артериальной гипертонией (АД 190/110 мм рт. ст.), нарушением зрения, анемией (содержание гемоглобина 69 г/л) и увеличением активности ЛДГ (847 ЕД/л). Количество тромбоцитов оставалось в пределах нормы (265-165 × 109/л). При иммунологическом исследовании данных за антифосфолипидный синдром, системную красную волчанку и АНЦА-ассоциированный васкулит не выявлено. Учитывая сочетание ОПП с признаками микроангиопатического гемолиза, обсуждалась ТМА, хотя отсутствие тромбоцитопении не укладывалось в этот диагноз. Для уточнения характера поражения почек в феврале 2019 г. была выполнена пункционная биопсия почки: в препарате 10 клубочков, три из которых полностью склерозированы, в четырех клубочках просвет капиллярных петель практически полностью отсутствует из-за диффузного тромбоза (в одном клубочке) и набухания эндотелиальных клеток (в трех клубочках) с накоплением воспалительных и “пенистых" клеток в просвете капиллярных петель, диффузный склероз интерстиция и атрофия канальцев, занимающие более 50% площади паренхимы, просвет артерий среднего и малого колибра резко сужен за счет расширения субэндотелиального пространства и миоинтимальной пролиферации, в артериолах мукоидное набухание интимы. При иммунофлюоресцентном исследовании выявлено гранулярное свечение С3 в мезангии и на периферии. Гистологическая картина соответствовала ТМА. Для исключения ТТП определена активность ADAMTS-13, которая оказалась нормальной.
Пациенту проводилось лечение программным гемодиализом, однако состояние его оставалось тяжелым: сохранялись слабость, тошнота, одышка, отеки, повышение АД до 220/110 мм рт. ст. несмотря на прием 5 антигипертензивных препаратов. При обследовании в клинике им. Е.М. Тареева в марте 2019 г. выявлены МАГА (гемоглобин 79 г/л, увеличение активности ЛДГ до 449 ЕД/л), гиперкреатининемия (1173 мкмоль/л), снижение содержания С3 компонента комплемента, отрицательная проба Кумбса. Количество тромбоцитов составляло 308 × 109/л. Кроме того, определялись признаки поражения сердца (снижение фракции выброса левого желудочка до 49%) и органа зрения (пурчероподобная ретинопатия).
Учитывая развитие у пациента ТМА, подтвержденной гистологически, в сочетании с нормальной активностью ADAMTS-13 и снижением содержания С3 компонента комплемента, был диагностирован аГУС, триггером которого, по-видимому, послужила бронхолегочная инфекция. После вакцинации противоменингококковой вакциной в мае 2019 г. была начата терапия экулизумабом, которая привела к купированию гемолиза (гемоглобин – 119 г/л, ЛДГ – 280 ЕД/л), стабилизации АД, появлению диуреза (до 1 л/сут), значительному улучшению состояния, в том числе уменьшению слабости и одышки, увеличению толерантности к физической нагрузке. Через полгода фракция выброса левого желудочка увеличилась до 69%, полностью восстановилась острота зрения, стабилизировались гемодинамические показатели (АД контролируется приемом амлодипина 10 мг/сут на уровне 130/85 мм рт. ст.), хотя, несмотря на появление диуреза, функция почек не восстановилась, а пациент остается диализзависимым. При молекулярно-генетическом исследовании выявлена патогенная мутация гена фактора Н (c.2596+1G>T), ассоциированная с развитием аГУС. В настоящее время пациент продолжает терапию экулизумабом, клинико-лабораторные признаки активности ТМА отсутствуют, планируется аллотрансплантация почки.
К.А. Демьянова. Развитие ОПП может быть обусловлено различными причинами, включая как специфические заболевания почек (например, острый интерстициальный нефрит, острые гломерулярные и сосудистые поражения почек), неспецифические состояния (например ишемия, токсическое повреждение), а также экстраренальные нарушения (например, преренальная азотемия и острая постренальная обструктивная нефропатия) [11]. Причиной острого ухудшения функции почек могут быть антифосфолипидный синдром и быстрогрессирующий гломерулонефрит, который обычно развивается при АНЦАассоциированных васкулитах и болезни, обусловленной антителами к базальной мембране клубочка (анти-БМК болезнь, или синдром Гудпасчера). Однако в случае развития ОПП у молодых пациентов с гематологическими нарушениями и тяжелой артериальной гипертонией в первую очередь необходимо исключать ТМА. В нашем наблюдении сочетание ОПП с признаками микроангиопатического гемолиза (анемия, отрицательная проба Кумбса, повышение активности ЛДГ) при отсутствии иммунологических маркеров системной патологии свидетельствовало о наличии ТМА, диагноз которой был подтвержден при биопсии почки. У пациентов с ТМА диагноз аГУС может быть установлен только при отсутствии ТТП и STEC-ГУС, т.е. других первичных ТМА. Диагноз ТТП был исключен на основании определения активности ADAMTS-13, оказавшейся ожидаемо нормальной, поскольку тромбоцитопении, прямо коррелирующей с активностью этой протеиназы, у пациента не было. При STEC-ГУС, который является самым распространенным вариантом гемолитико-уремического синдрома, прежде всего у детей, в крови и кале выявляют шига-токсин, продуцируемый E. coli [6], однако мы не проводили соответствующие исследования, учитывая возраст пациента и отсутствие у него диареи и других признаков поражения желудочно-кишечного тракта. Окончательно диагноз аГУС был подтвержден наличием патогенной мутации гена фактора Н (c.2596+1G>T), ассоциированной с развитием этого заболевания. Как указано выше, триггером аГУС примерно у половины пациентов (и еще чаще у детей) являются различные комплемент-активирующие состояния, в частности у нашего пациента таковым была бронхолегочная инфекция.
С.В. Моисеев. Какую роль играет биопсия почки в диагностике ТМА? Нужно ли проводить ее всем больным, у которых обсуждается этот диагноз? Насколько необходимы исследование компонентов системы комплемента и молекулярно-генетические тесты для подтверждения диагноза аГУС?
Н.Л. Козловская. Пункционную биопсию почки не считают обязательным методом исследования, необходимым для подтверждения диагноза ТМА [4], однако она обоснована, если этот диагноз по каким-то причинам вызывает сомнение, особенно при отсутствии полного симптомокомплекса ТМА. Пункционная биопсия почки, как и любое другое инвазивное исследование, представляет определенную опасность, так как она может вызвать дополнительную активацию системы комплемента. В представленном наблюдении основанием для биопсии почки послужило отсутствие тромбоцитопении, которая, наряду с ОПП и МАГА, входит в триаду классических проявлений ТМА. Следует отметить достаточно высокую частоту неполного лабораторного симптомокомплекса ТМА у пациентов с аГУС, в первую очередь, отсутствие тромбоцитопении, что однако не исключает этот диагноз. Так, в одном исследовании количество тромбоцитов в крови было нормальным у 44% из 50 больных с гистологически подтвержденной ТМА [12], а в нашей выборке тромбоцитопения отсутствовала у 12 (14%) из 85 больных неакушерским аГУС [13].
Исследование большинства компонентов комплемента и факторов, регулирующих активность этой системы, возможно в рамках научных исследований, в то время как в обычной клинической практике доступно только определение содержания С3 и С4 компонентов комплемента, реже – общей гемолитической активности комплемента СН50. Чувствительность этих показателей в диагностике комплемент-опосредованной ТМА низкая, в частности снижение содержания С3 компонента комплемента при нормальном уровне С4 наблюдается не более, чем у половины больных с аГУС. Тем не менее, снижение содержания С3 у нашего пациента послужило дополнительным аргументом в пользу этого диагноза. У пациентов с аГУС чаще всего определяются мутации гена фактора H, которая была выявлена и в представленном наблюдении. Проведение молекулярно-генетического исследования не считают необходимым для подтверждения диагноза аГУС, так как мутации генов регуляторных белков АПК обнаруживают только у 60-70% больных с наследственным аГУС и 30% больных со спорадической формой болезни, поэтому их отсутствие не исключает диагноз комплемент-опосредованной ТМА [4]. Кроме того, для выполнения генетического исследования обычно требуется не менее 2 месяцев, в то время как начинать лечение ТМА следует как можно раньше, учитывая высокий риск смерти или необратимого повреждения почек и других органов при несвоевременно назначенной терапии. Однако риск рецидива аГУС после трансплантации почки зависит от вида мутаций (75-90% при наличии мутации гена фактора H), поэтому при подготовке к трансплантации почки в план обследования пациента с аГУС необходимо обязательно включать молекулярно-генетическое исследование [4].
С.В. Моисеев. Ведущим в клинической картине аГУС является поражение почек, которое часто приводит к развитию терминальной хронической почечной недостаточности, требующей заместительной почечной терапии. Какие еще органы поражаются при этом заболевании?
Ю.В. Коротчаева. Комплемент-опосредованная ТМА – это системное заболевание, которое сопровождается тромбообразованием в сосудах микроциркуляторного русла не только почек, но и других органов. По данным международного регистра, в который включены более 550 пациентов с аГУС, признаки поражения сердца отмечались у 31,6% больных, желудочно-кишечного тракта – у 37,4%, нервной системы – у 29,9% и легких – у 14,0% [14]. У представленного нами пациента наблюдались поражение сердца, характеризовавшееся снижением фракции выброса левого желудочка, и пурчероподобная ретинопатия, которая обычно развивается после травмы черепа, но описана и при многих других заболеваниях, включая аГУС [15]. Следует отметить, что сократительной функция левого желудочка и острота зрения быстро восстановились после начала лечения экулизумабом. Возможность тяжелого поражения разных органов при аГУС демонстрирует следующее наблюдение.
М.В. Алексеева. Пациентка М., 27 лет. В возрасте 23 лет первая беременность протекала без осложнений и закончилась самостоятельными родами. На 30-й неделе настоящей беременности отмечены повышение АД до 140/90 мм рт. ст. и появление протеинурии 1 г/л. Была диагностирована преэклампсия, назначена антигипертензивная терапия, однако на 35-й неделе беременности протеинурия увеличилась до 8 г/л, а АД – до 180/100 мм рт. ст. Выполнено экстренное оперативное родоразрешение, осложнившееся атоническим кровотечением, для купирования которого потребовались повторная лапаротомия и гистерэктомия. В течение суток после родоразрешения отмечено ухудшение состояния пациентки с развитием анурии, эпизода тонико-клонических генерализованных судорог с потерей сознания, нарастающей дыхательной недостаточностью, потребовавшей вентиляционной поддержки. По данным магнитно-резонансной томографии головного мозга определялась картина острой ишемии в теменно-затылочных областях с двух сторон. При обследовании выявлены анемия (гемоглобин – 69 г/л), тромбоцитопения (22 × 109/л), повышение сывороточного уровня креатинина (359 мкмоль/л), активности печеночных аминотрасфераз (АСТ – 210 ЕД/л, АЛТ – 232 ЕД/л) и ЛДГ (1170 ЕД/л). Учитывая сочетание ОПП с МАГА и тромбоцитопенией, а также поражением печени, легких и ЦНС, была диагностирована ТМА. Ухудшение состояния после родоразрешения позволило исключить преэклампсию и HELLP-синдром как причины ТМА. Данных в пользу ТТП (активность ADAMTS-13 – 61%), катастрофического антифосфолипидного синдрома и системной красной волчанки (отсутствие антифосфолипидных антител, антител к ДНК и антинуклеарного фактора) также не выявлено, в связи с чем установлен диагноз аГУС. Проводимая с первого дня плазмотерапия в режиме плазмообмена не привела к улучшению состояния пациентки. На 4-й день начата терапия экулизумабом. Уже на 3-й день после начала лечения появился диурез до 1,5 л/сут, восстановилось сознание, регрессировала неврологическая симптоматика, нормализовалось число тромбоцитов (160 × 109/л). Были прекращены гемодиализ и искусственная вентиляция легких. Через 2 недели нормализовались все клинические и биохимические лабораторные показатели. У пациентки полностью восстановилась функция почек. Через 3 месяца при молекулярно-генетическом исследовании патогенных мутаций генов системы комплемента, ассоциированных с аГУС, не выявлено, а терапия экулизумабом была прекращена. В настоящее время, спустя 3 года, сохраняется ремиссия заболевания.
Ю.В. Коротчаева. Настоящее наблюдение демонстрирует развитие тяжелого аГУС, проявлявшегося не только ОПП, но и поражением легких, центральной нервной системы и печени, у пациентки без патогенных мутаций генов системы комплемента. Триггерами активации системы комплемента в данном случае, кроме самой беременности, были ее осложнения, в том числе преэклампсия, кровотечение и повторные оперативные вмешательства (кесарево сечение, релапаротомия, гистерэктомия). Все они представляют собой комплемент-активирующие состояния, которые развились в короткий промежуток времени и оказали содружественное действие на систему комплемента, что и привело к развитию аГУС несмотря на отсутствие патогенных мутаций генов системы комплемента [16]. На долю акушерского аГУС приходится около 7% всех случаев аГУС в популяции и до 20% случаев у женщин [17]. По данным французского когортного исследования, беременность является ведущей причиной вторичной ТМА (35%) [18]. Клинические исследования позволили предположить, что акушерский аГУС по своей структуре может быть неоднородным и включать как “классический" аГУС, так и “вторичный" аГУС, не связанный с конституциональной дисрегуляцией системы комплемента, но сопоставимый по тяжести и неблагоприятному прогнозу [19]. По данным международного регистра, частота патогенных мутаций генов системы комплемента и антител к фактору Н была сходной у женщин с акушерским и неакушерским аГУС (43% и 45%, соответственно). Доля пациенток с семейным анамнезом заболевания также была сопоставимой в этих двух группах [17]. Акушерский аГУС сопровождается высокими перинатальной и материнской заболеваемостью и смертностью, в частности частым развитием терминальной хронической почечной недостаточности, требующей заместительной почечной терапии, в исходе неразрешившегося ОПП. Особенностями акушерского аГУС являются тяжелое и быстропрогрессирующее течение и частое поражение жизненно-важных органов (помимо почек) с развитием полиорганной недостаточности), обусловленной быстрой генерализацией тромбообразования в сосудах микроциркуляторного русла [20]. Следует отметить, что не все исследования подтверждают эту точку зрения. Так, в международном регистре тяжесть поражения почек и частота поражения других органов были сходными в группах женщин с акушерским и неакушерским аГУС [17].
С.В. Моисеев. Приведенные случаи демонстрируют эффективность экулизумаба в лечении аГУС. Особенно впечатляющими оказались результаты терапии у пациентки с акушерским аГУС, у которой в течение короткого срока после назначения препарата удалось добиться выздоровления, в том числе полного восстановления функции почек и легких, что позволило прекратить заместительную почечную терапию и респираторную поддержку. Каковы современные подходы к лечению аГУС? Всем ли больным следует назначать экулизумаб? Как долго продолжать терапию этим препаратом?
Ю.В. Коротчаева. Эффективность экулизумаба у первого пациента также не вызывает сомнения. При ме нение ингибитора комплемента позволило купироватьмикроангиопатический гемолиз и привело к снижениюАД, нормализации сократительной функции левогожелудочка и остроты зрения, хотя функция почек невосстановилась, а пациент остается зависимым от диализа. Разница результатов лечения у двух пациентовотражает неодинаковые сроки инициации комплементблокирующей терапии. Пациентке с акушерским аГУСс первого дня проводилась плазмотерапия, а экулизумаб был назначен на 4-й день после появления признаков ТМА, в то время как у первого пациента лечениеингибитором комплемента было начато только через 4месяца после дебюта заболевания в связи с позднейдиагностикой аГУС. Наши собственные данные свидетельствуют о том, что отсроченное начало патогенетической терапии (более 3 недель) уменьшает шанс навосстановление функции почек почти в 50 раз [21]. Приэтом отсутствие патогенных мутаций генов комплемента у пациентки с акушерским аГУС позволило с высокой вероятностью исключить риск рецидива аГУС ичерез несколько месяцев прекратить комплемент-блокирующую терапию. Как указано выше, у пациентов смутациями гена фактора H риск рецидива заболевания,в частности после трансплантации почки, наоборот,очень высокий, поэтому в таких случаях необходимопродолжать лечение экулизумабом.
Н.Л. Козловская. На протяжении многих лет для лечения любой недифференцированной ТМА применяется плазмотерапия, которую предпочтительно проводить в режиме плазмообмена, позволяющего вводить большие объемы свежезамороженной плазмы без развития гиперволемии и гипергидратации [4]. Введение свежезамороженной плазмы позволяет устранить дефицит регуляторов активности системы комплемента и подавить тромбообразование в сосудах микроциркуляторного русла под действием компонентов плазмы, обладающих антикоагулянтной и фибринолитической активностью. При плазмообмене удаляются также циркулирующие ингибиторы системы комплемента и антитела к фактору H. Однако эффективность плазмотерапии при аГУС ограничена преимущественно влиянием на картину крови и неопределенной продолжительностью эффекта, поэтому при отсутствии ответа на лечение в течение первой недели или развитии нежелательных явлений необходимо назначать экулизумаб – гуманизированные моноклональные антитела к С5 компоненту комплемента. Экулизумаб блокирует расщепление С5 и образование анафилатоксина С5а и мембраноатакующего комплекса С5b-C9, поэтому его применение предотвращает повреждение эндотелия сосудов и подавляет тромбообразование в сосудах микроциркуляторного русла. Блокада каскада активации комплемента на конечном этапе дает возможность избежать нарушения активации “проксимальной" части системы комплемента (С1, С2, С3, С4), которая выполняет важные защитные функции.
Первоначально экулизумаб применялся для лечения пароксизмальной ночной гемоглобинурии [22], а позднее он был зарегистрирован и для лечения комплементопосредованной ТМА на основании результатов двух 26-недельных открытых, неконтролируемых исследований в целом у 34 больных [23]. В первое исследование включали пациентов с прогрессирующей ТМА, которым было проведено по крайней мере 4 сеанса обмена или трансфузии плазмы за неделю до скрининга. Во втором исследовании принимали участие пациенты с длительным анамнезом аГУС (медиана 48,3 месяца) и хронической почечной недостаточностью, у которых число тромбоцитов было стабильным на фоне трансфузий или обмена плазмы. В первом исследовании лечение экулизумабом привело к значительному увеличению числа тромбоцитов в среднем на 73 × 109/л через 26 недель (p<0,001) и на 91 × 109/л через 64 недели (p<0,001). У 87% пациентов число тромбоцитов оставалось нормальным через 26 и 64 недели. Во втором исследовании доля пациентов, у которых отсутствовали проявления ТМА, составила 80% через 26 недель и достигла 85% через 62 недели. В обоих исследованиях терапия экулизумабом вызывала достоверное улучшение функции почек.
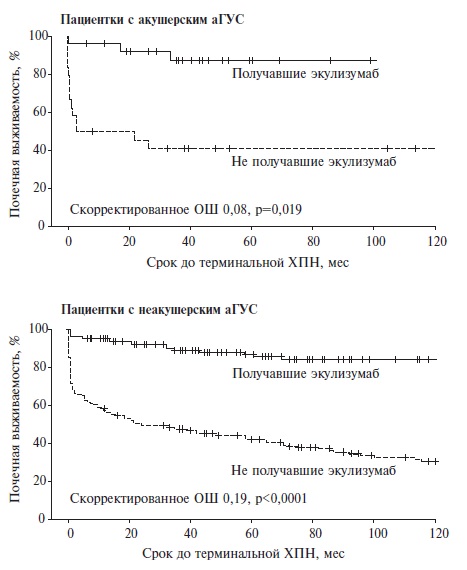
В настоящее время эффективность и безопасность экулизумаба продолжают изучаться в когортных исследованиях, в том числе международном регистре пациентов с аГУС. Недавно F. Fakhouri и соавт. оценили почечные исходы заболевания у 51 женщины с акушерским аГУС и 397 пациенток репродуктивного возраста, у которых триггеры аГУС не были выявлены [17]. Лечение экулизумабом в этих двух группах проводилось примерно у половины больных (у 27 и 187, соответственно). По данным регрессионного анализа Кокса с поправкой на различные факторы, способные повлиять на исходы аГУС, лечение экулизумабом привело к достоверному снижению риска развития терминальной хронической почечной недостаточности у пациенток как с акушерским, так и неакушерским аГУС (рис. 2). В обеих группах при лечении ингибитором комплемента было выявлено увеличение средней расчетной скорости клубочковой фильтрации на 56,2±39,8 и 40,9±32,1 мл/мин/1,73 м2, соответственно. Наши данные у 76 пациенток с акушерским аГУС и 85 взрослых больных с неакушерским аГУС также подтверждают эффективность экулизумаба [13]. Так, среди пациенток с акушерским аГУС восстановление или улучшение функции почек было достигнуто у 78,3% и 56,6% женщин, получавших и не получавших экулизумаб, а среди больных с неакушерским аГУС – у 52,9% и 33,8%, соответственно.
С.В. Моисеев. В России разработан первый биоаналог экулизумаба, который в 2019 г. был зарегистрирован для лечения пароксизмальной ночной гемоглобинурии и аГУС под торговым названием Элизария. Соответственно, сегодня в нашей стране все пациенты с ранее диагностированным и впервые выявленным аГУС получают лечение этим препаратом. Можно ли судить о сравнительной эффективности оригинального экулизумаба и его биоаналога?
Ю.В. Коротчаева. В рандомизированных контролируемых исследованиях у пациентов с аГУС эти два препарата не сравнивали, однако такие исследования проводились у пациентов с ночной пароксизмальной гемоглобинурией [24-26]. Клинический опыт также свидетельствует о том, что биоаналог не уступает оригинальному экулизумабу по эффективности и безопасности как у детей, так и взрослых [27-29]. В ретроспективном исследовании мы оценили результаты применения экулизумаба у 50 пациенток с акушерским аГУС, 9 из которых получали биоаналог, а 41 – оригинальный препарат (18 из них в 2019 г. были переведены на терапию биоаналогом) [30]. Пациентки в двух группах исходно не различались по возрасту, акушерскому анамнезу, количеству комплемент-активирующих состояний и основным клинико-лабораторным параметрам в острый период заболевания. В результате лечения гематологические показатели нормализовались во всех случаях, а функция почек полностью восстановилась у 88,9% и 80,5% пациенток, соответственно. Как и следовало ожидать, почечные исходы были значительно лучше, если терапию ингибитором комплемента начинали в течение первых 2 недель от дебюта заболевания. Ни у одной из 18 пациенток, которые были переведены с оригинального экулизумаба на биоаналог, не было отмечено рецидива ТМА. Полученные данные свидетельствуют о взаимозаменяемости оригинального экулизумаба и его отечественного биоаналога.
С.В. Моисеев. Возможно ли прекращение терапии экулизумабом у пациентов с аГУС?
Н.Л. Козловская. Первоначально предполагалось, что пациентам с аГУС показана пожизненная терапия экулизумабом, учитывая генетическую природу аГУС. Однако сегодня отношение к продолжительности комплемент-блокирующей терапии при этом заболевании меняется, поскольку оказалось, что в ряде случаев препарат может быть отменен после достижения стойкой ремиссии заболевания. Общепризнано, что вопрос отмены следует решать индивидуально, так как прекращение комплемент-блокирующей терапии может привести к рецидиву ТМА. G. Ariceta и соавт. изучили последствия отмены экулизумаба у 151 пациента с ТМА, ответившего на лечение этим препаратом [31]. Медиана длительности лечения ингибитором комплемента составила 0,96 лет, а медиана длительности наблюдения – 2,3 года. У 33 (21,9%) пациентов развился рецидив ТМА в течение в среднем 5,3 мес после отмены препарата, в связи с чем лечение в большинстве случаев было возобновлено. По данным многофакторного анализа, факторами риска рецидива ТМА были наличие патогенной мутации генов системы комплемента и семейный анамнез аГУС. Кроме того, у 12 (8%) больных отмечено развитие терминальной хронической почечной недостаточности, потребовавшей заместительной почечной терапии, а у 40 (27%) – внепочечных проявлений аГУС.
В проспективном многоцентровом французском исследовании у 51 пациента с аГУС, ответившего на лечение экулизумабом, отмена этого препарата привела к рецидиву ТМА в 13 (23%) случаях [32]. Факторами риска рецидива были женский пол и наличие редких мутаций генов системы комплемента. У 11 из 13 больных после возобновления терапии экулизумабом функция почек улучшилась, однако в 1 случае авторы наблюдали развитие терминальной хронической почечной недостаточности. Тем не менее, исследователи сделали вывод о возможности прекращения комп лемент-блокирующей терапии у части больных на основании результатов молекулярно-генетического исследования.
В целом приведенные данные свидетельствуют о том, что риск развития рецидива ТМА у пациентов с аГУС достаточно высокий, причем прекращение комплементблокирующей терапии в части случаев приводит к серьезным последствиям, в том числе прогрессированию хронической почечной недостаточности или развитию внепочечных проявлений заболевания. Однако мы не исключаем возможность отмены экулизумаба в случае достижения ремиссии аГУС при наличии некоторых мутаций генов комплемента, которые ассоциируются с низким риском рецидива ТМА, или при отсутствии патогенных мутаций генов этой системы. Так, у второй пациентки, у которой аГУС развился под действием осложнений беременности, а мутации генов комплемента выявлены не были, экулизумаб был отменен через несколько месяцев после купирования всех проявлений ТМА. При этом в течение 3 лет наблюдения рецидивов заболевания не отмечено. После прекращения терапии экулизумабом необходимо регулярно контролировать лабораторные показатели, в частности содержание гемоглобина и сывороточного креатинина, количество тромбоцитов в крови и активность ЛДГ, чтобы своевременно диагностировать рецидив ТМА и возобновить лечение.
С.В. Моисеев. Представленные наблюдения иллюстрируют проявления аГУС – ОПП, которое сочетается с МАГА и тромбоцитопенией, а также основные триггеры этого заболевания и эффективность экулизумаба. Необходимо еще раз подчеркнуть важность ранней диагностики аГУС, так как своевременно начатая патогенетическая терапия обеспечивает не только нормализацию гематологических показателей, но и позволяет значительно улучшить или полностью восстановить функцию почек и, соответственно, прекратить лечение гемодиализом.
Используемые источники
- Campistol JM, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia 2015;35(5):421–47.
- Fakhouri F, Zuber J, Fromeaux-Bacchi V, Loirat C. Haemolytic uraemic syndrome. Lancet 2017;390:681–96.
- Yan K, Desai K, Gullapalli L, et al. Epidemiology of atypical hemolytic uremic syndrome: A systematic literature review. Clin Epidemiol 2020;12:295-305.
- Козловская Н.Л., Прокопенко Е.И., Эмирова Х.М., Серикова С.Ю. Клинические рекомендации по диагностике и лечению атипичного гемолитико-уремического синдрома. Нефрология и диализ 2015;17(3):242-64 [Kozlovskaya NL, Prokopenko EI, Emirova KhM, Serikova SYu. Clinical guidelines for diagnosis and treatment of atypical hemolytic uremic syndrome. Nephrology and Dialysis 2015;17(3):242-64 (In Russ.)].
- Noris M, Caprioli J, Bresin E, Mossali C, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010;5:1844–59.
- Козловская Н.Л. Атипичный гемолитико-уремический синдром: современные представления о патогенезе, клинике, подходах к диагностике и лечению. Тромбоз, гемостазиреология 2019;80(4):13-20 [Kozlovskaya NL. Atypical hemolytic uremic syndrome: current understanding of the pathogenesis, clinic, approaches to diagnosis and treatment. Tromboz, gemostaz i reologiya 2019;80(4):13-20 (In Russ.)].
- Goodship TH, Cook HT, Fakhouri F, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a "Kidney Disease: Improving Global Outcomes" (KDIGO) Controversies Conference. Kidney Int 2017;91(3):539-51.
- Praga M, Rodríguez de Córdoba S. Secondary atypical hemolytic uremic syndromes in the era of complement blockade. Kidney Int 2019;95(6):1298-300.
- Yoshida Y, Kato H, Ikeda Y, Nangaku M. Pathogenesis of atypical hemolytic uremic syndrome. J Atheroscler Thromb 2019;26(2):99-110.
- Алтынова В.Х., Эмирова Х.М., Нигматуллина Н.Б. и др. Атипичный гемолитико-уремический синдром в педиатрической практике. Клин фармакол тер 2016;25(3):83-9 [Altynova VKh, Emirova KhM, Nigmatullina NB, et al. Аtypical hemolytic uremic syndrome in pediatric patient. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2016;25(3):83-9 (In Russ.)].
- Kellum JA, Romagnani P, Ashuntantang G, et al. Acute kidney injury. Nat Rev Dis Primers 2021 Jul 15;7(1):52.
- De Serres SA, Isenring P. Athrombocytopenic thrombotic microangiopathy, a condition that could be overlooked based on current diagnostic criteria. Nephrol Dial Transplant 2009;24(3):1048-50.
- Козловская Н.Л., Коротчаева Ю.В., Демьянова К.А., Шифман Е.М. Сравни тельная характеристика акушерского и “общепопуляционного” атипичного гемолитико-уремического синдрома у взрослых. Нефрология и диализ 2022;24(1):52-61 [Kozlovskaya NL, Korotchaeva YV, Demyanova K.A., Shifman E.M. Comparative characteristics of obstetric and “population-wide” atypical hemolytic-uremic syndrome in adults. Nephrologу and Dialуsis 2022;24(1):52-61 (In Russ.)].
- Greenbaum LA, Licht C, Nikolaou V, et al. Functional assessment of fatigue and other patient-reported outcomes in patients enrolled in the Global aHUS Registry. Kidney Int Rep 2020;5(8):1161-71.
- Gange WS, Haghighi A, Toy BC. Purtscher-like retinopathy associated with atypical hemolytic uremic syndrome: case report and review of outcomes. Retin Cases Brief Rep 2021 Jan 18.
- Коротчаева Ю.В., Козловская Н.Л., Демьянова К.А. и др. Генетические аспекты акушерского атипичного гемолитико-уремического синдрома. Клиническая нефрология 2017;1:12-7 [Korotchaeva YV, Kozlovskaya NL, Demyanova KA, et al. Genetic aspects of obstetric atypical hemolytic uremic syndrome. Clinical Nephrology 2017;1:12-7 (In Russ.)].
- Fakhouri F, Scully M, Ardissino G, et al.·Pregnancy-triggered atypical hemolytic uremic syndrome (aHUS): a Global aHUS Registry analysis. J Nephrol 2021;34:1581–90.
- Bayer G, von Tokarski F, Thoreau B, et al. Etiology and outcomes of thrombotic microangiopathies. Clin J Am Soc Nephrol 2017;14(4):557.
- Коротчаева Ю.В., Козловская Н.Л., Шифман Е.М., Демьянова К.А. "Поздние" осложнения беременности как триггеры акушерского атипичного гемолитико-уремического синдрома. Нефрология и диализ 2020;22(2):198-209 [Korotchaeva YV, Kozlovskaya NL, Shifman EM, Demyanova KA. Late pregnancy complications as a triggers of obstetric atypical hemolytic uremic syndrome. Nephrology and Dialysis 2020 22(2):198-209 (In Russ.)].
- Kozlovskaya NL, Korotchaeva YV, Bobrova LA. Adverse outcomes in obstetricatypical haemolytic uraemic syndrome: a case series analysis. J Matern Fetal Neonat Med 2019;32(17):2853-9.
- Козловская Н.Л., Коротчаева Ю.В., Шифман Е.М., Боброва Л.А. Атипичный гемолитико-уремический синдром как одна из причин острого повреждения почек у беременных. Терапевтический архив 2018;90(6):28-32 [Kozlovskaya NL, Korotchaeva YV, Shifman EM, Bobrova LA. Atypical hemolytic-uremic syndrome as one of the causes of acute kidney injury in pregnant women. Terapevticheskii arkhiv 2018;90(6):28-34 (In Russ.)].
- Rother RP, Rollins SA, Mojcik CF, et al. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat Biotechnol 2007;25(11):1256–64.
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med 2013;368:2169-81.
- Kulagin A, Ptushkin V, Lukina E, et al. Phase III clinical trial of Elizaria® and Soliris® in adult patients with paroxysmal nocturnal hemoglobinuria: results of comparative analysis of efficacy, safety, and pharmacological data. Blood 2019; 134(Suppl 1):3748.
- Kulagin AD, Ptushkin VV, Lukina EA, et al. Randomized multicenter noninferiority phase III clinical trial of the first biosimilar of eculizumab. Ann Hematol 2021;100(11):2689–98.
- Птушкин В.В., Кулагин А.Д., Лукина Е.А. и др. Результаты открытого многоцентрового клинического исследования Ib фазы по оценке безопасности, фармакокинетики и фармакодинамики первого биоаналога экулизумаба у нелеченных пациентов с пароксизмальной ночной гемоглобинурией в фазе индукции терапии. Терапевтический архив 2020;92(7):77-84 [Ptushkin VV, Kulagin AD, Lukina EA, et al. Results of phase Ib open multicenter clinical trial of the safety, pharmacokineticsand pharmacodynamics of first biosimilar of eculizumab in untreated patients with paroxysmal nocturnal hemoglobinuria during induction of therapy. Therapeutic Archive 2020;92(7):77–84 (In Russ.)].
- Эмирова Х.М., Орлова О.М., Музуров А.Л. и др. Опыт применения Элизарии® при атипичном гемолитико-уремическом синдроме. Педиатрия 2019;98(5):225-9 [Emirova KhМ, Orlova OM, Мuzurov АL, et al. The experience of using Elizaria® for atypical hemolytic uremic syndrome. Pediatria 2019;98(5):225-9 (In Russ.)].
- Лаврищева Ю.В., Яковенко А.А., Кудлай Д.А. Опыт применения российского биоаналога оригинального препарата экулизумаба для лечения больных атипичным гемолитико-уремическим синдромом. Терапевтический архив 2020;92(6):76-80 [Lavrishcheva IuV, Jakovenko AA, Kudlay DA. The experience of using the Russian biosimilar of the original drug eculizumab for the treatment of patients with atypical hemolytic-uremic syndrome. Therapeutic Archive 2020;92(6):76–80 (In Russ.)].
- Lavrishcheva I.V., Jakovenko A.A., Kudlay D.A. A case report of atypical hemolytic-uremic syndrome treatment with the first Russian eculizumab in adult patient. Urol Nephrol 2020;8(2):37-40.
- Коротчаева Ю.В., Козловская Н.Л., Шифман Е.М. Сравнительный анализ эффективности препаратов экулизумаба в лечении акушерского атипичного гемолитико-уремического синдрома. Клин фармакол тер 2021;30(3):25-30 [Korotchaeva YV, Kozlovskaya NL, Shifman EM. Comparative efficacy of the original and biosimilar eculizumab in the treatment of obstetric atypical hemolytic-uremic syndrome. Klinicheskaya farmakologiya i terapiya = Clin Pharmacol Ther 2021;30(3):25-30 (In Russ.)].
- Ariceta G, Fakhouri F, Sartz L, et al. Eculizumab discontinuation in atypical haemolytic uraemic syndrome: TMA recurrence risk and renal outcomes. Clin Kidney J 2021;14(9):2075-84.
- Fakhouri F, Fila M, Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicentric study. Blood 2021;137(18):2438-49.